
Latest recommendations

| Id | Title | Authors | Abstract▲ | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
13 Jan 2019
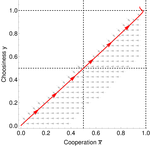
Why cooperation is not running awayA nice twist on partner choice theoryRecommended by Erol Akcay based on reviews by 2 anonymous reviewersIn this paper, Geoffroy et al. [1] deal with partner choice as a mechanism of maintaining cooperation, and argues that rather than being unequivocally a force towards improved payoffs to everyone through cooperation, partner choice can lead to “over-cooperation” where individuals can evolve to invest so much in cooperation that the costs of cooperating partially or fully negate the benefits from it. This happens when partner choice is consequential and effective, i.e., when interactions are long (so each decision to accept or reject a partner is a bigger stake) and when meeting new partners is frequent when unpaired (so that when one leaves an interaction one can find a new partner quickly). Geoffroy et al. [1] show that this tendency to select for overcooperation under such regimes can be counteracted if individuals base their acceptance-rejection of partners not just on the partner cooperativeness, but also on their own. By using tools from matching theory in economics, they show that plastic partner choice generates positive assortment between cooperativeness of the partners, and in the extreme case of perfectly assortative pairings, makes the pair the unit of selection, which selects for maximum total payoff. References [1] Geoffroy, F., Baumard, N., & Andre, J.-B. (2019). Why cooperation is not running away. bioRxiv, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/316117 | Why cooperation is not running away | Félix Geoffroy, Nicolas Baumard, Jean-Baptiste André | <p>A growing number of experimental and theoretical studies show the importance of partner choice as a mechanism to promote the evolution of cooperation, especially in humans. In this paper, we focus on the question of the precise quantitative lev... | | Behavior & Social Evolution, Evolutionary Theory | Erol Akcay | 2018-05-15 10:32:51 | ||
16 Mar 2023
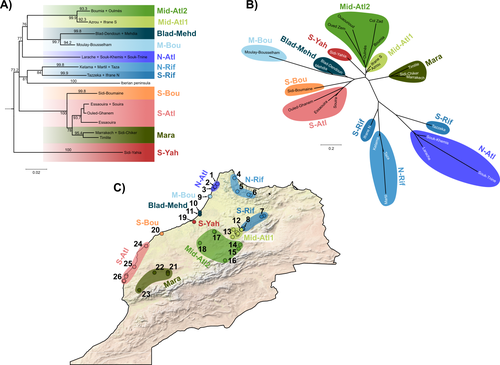
Phylogeographic breaks and how to find them: Separating vicariance from isolation by distance in a lizard with restricted dispersalThe difficult task of partitioning the effects of vicariance and isolation by distance in poor dispersersRecommended by Eric Pante based on reviews by Kevin Sánchez and Aglaia (Cilia) Antoniou based on reviews by Kevin Sánchez and Aglaia (Cilia) Antoniou
Partitioning the effects of vicariance and low dispersal has been a long-standing problem in historical biogeography and phylogeography. While the term “vicariance” refers to divergence in allopatry, caused by some physical (geological, geographical) or climatic barriers (e.g. Rosen 1978), isolation by distance refers to the genetic differentiation of remote populations due to the physical distance separating them, when the latter surpasses the scale of dispersal (Wright 1938, 1940, 1943). Vicariance and dispersal have long been considered as separate forces leading to separate scenarii of speciation (e.g. reviewed in Hickerson and Meyer 2008). Nevertheless, these two processes are strongly linked, as, for example, vicariance theory relies on the assumption that ancestral lineages were once linked by dispersal prior to physical or climatic isolation (Rosen 1978). Low dispersal and vicariance are not mutually exclusive, and distinguishing these two processes in heterogeneous landscapes, especially for poor dispersers, remains therefore a severe challenge. For example, low dispersal (and/or small population size) can give rise to geographic patterns consistent with a phylogeographic break and be mistaken for geographic isolation (Irwin 2002, Kuo and Avise 2005). The study of Rancilliac and colleagues (2023) is at the heart of this issue. It focuses on a nominal lizard species, the red-tailed spiny-footed lizard (Acanthodactylus erythrurus, Squamata: Lacertidae), which has a wide spatial distribution (from the Maghreb to the Iberian Peninsula), is found in a variety of different habitats, and has a wide range of morphological traits that do not always correlate with phylogeny. The main question is the following: have “the morphological and ecological diversification of this group been produced by vicariance and lineage diversification, or by local adaptation in the face of historical gene flow?” To tackle this question, the authors used sequence data from multiple mitochondrial and nuclear markers and a nested analysis workflow integrating phylogeography, multiple correspondence analyses and a relatively novel approach to IBD testing (Hausdorf & Henning, 2020). The latter is based on regression analysis and was shown to be less prone to error than the traditional (partial) Mantel test. While this set of methods allowed the partitioning of the effect of isolation by distance and vicariance in shaping contemporary genetic diversity in red-tailed spiny-footed lizards, some of the evolutionary history of this species complex remains blurred by ongoing gene flow and admixture, retention of ancestral polymorphism, or selection. The lack of congruence between mitochondrial and nuclear gene trees once again warns us that proposing evolutionary scenarii based on individual gene trees is a risky business. References Hausdorf B, Hennig C (2020) Species delimitation and geography. Molecular Ecology Resources, 20, 950–960. https://doi.org/10.1111/1755-0998.13184 Hickerson MJ, Meyer CP (2008) Testing comparative phylogeographic models of marine vicariance and dispersal using a hierarchical Bayesian approach. BMC Evolutionary Biology, 8, 322. https://doi.org/10.1186/1471-2148-8-322 Irwin DE (2002) Phylogeographic breaks without geographic barriers to gene flow. Evolution, 56, 2383–2394. https://doi.org/10.1111/j.0014-3820.2002.tb00164.x Kuo C-H, Avise JC (2005) Phylogeographic breaks in low-dispersal species: the emergence of concordance across gene trees. Genetica, 124, 179–186. https://doi.org/10.1007/s10709-005-2095-y Rancilhac L, Miralles A, Geniez P, Mendez-Aranda D, Beddek M, Brito JC, Leblois R, Crochet P-A (2023) Phylogeographic breaks and how to find them: An empirical attempt at separating vicariance from isolation by distance in a lizard with restricted dispersal. bioRxiv, 2022.09.30.510256, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.09.30.510256 Rosen DE (1978) Vicariant Patterns and Historical Explanation in Biogeography. Systematic Biology, 27, 159–188. https://doi.org/10.2307/2412970 Wright, S (1938) Size of population and breeding structure in relation to evolution. Science 87:430-431. Wright S (1940) Breeding Structure of Populations in Relation to Speciation. The American Naturalist, 74, 232–248. https://doi.org/10.1086/280891 Wright S (1943) Isolation by distance. Genetics, 28, 114–138. https://doi.org/10.1093/genetics/28.2.114 | Phylogeographic breaks and how to find them: Separating vicariance from isolation by distance in a lizard with restricted dispersal | Loïs Rancilhac, Aurélien Miralles, Philippe Geniez, Daniel Mendez-Arranda, Menad Beddek, José Carlos Brito, Raphaël Leblois, Pierre-André Crochet | <p>Aim</p> <p>Discontinuity in the distribution of genetic diversity (often based on mtDNA) is usually interpreted as evidence for phylogeographic breaks, underlying vicariant units. However, a misleading signal of phylogeographic break can arise... | | Phylogeography & Biogeography, Population Genetics / Genomics, Speciation, Systematics / Taxonomy | Eric Pante | Kevin Sánchez | 2022-10-05 13:11:28 | |
18 Jun 2020
Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in betweenMolecular evolution through the joint lens of genomic and population processes.Recommended by Guillaume Achaz based on reviews by Benoit Nabholz and 1 anonymous reviewerIn their perspective article, F Pouyet and KJ Gilbert (2020), propose an interesting overview of all the processes that sculpt patterns of molecular evolution. This well documented article covers most (if not all) important facets of the recurrent debate that has marked the history of molecular evolution: the relative importance of natural selection and neutral processes (i.e. genetic drift). I particularly enjoyed reading this review, that instead of taking a clear position on the debate, catalogs patiently every pieces of information that can help understand how patterns we observed at the genome level, can be understood from a selectionnist point of view, from a neutralist one, and, to quote their title, from "everything in between". The review covers the classical objects of interest in population genetics (genetic drift, selection, demography and structure) but also describes several genomic processes (meiotic drive, linked selection, gene conversion and mutation processes) that obscure the interpretation of these population processes. The interplay between all these processes is very complex (to say the least) and have resulted in many cases in profound confusions while analyzing data. It is always very hard to fully acknowledge our ignorance and we have many times payed the price of model misspecifications. This review has the grand merit to improve our awareness in many directions. Being able to cover so many aspects of a wide topic, while expressing them simply and clearly, connecting concepts and observations from distant fields, is an amazing "tour de force". I believe this article constitutes an excellent up-to-date introduction to the questions and problems at stake in the field of molecular evolution and will certainly also help established researchers by providing them a stimulating overview supported with many relevant references. References [1] Pouyet F, Gilbert KJ (2020) Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between. arXiv:1909.11490 [q-bio]. ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. url:https://arxiv.org/abs/1909.11490 | Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between | Fanny Pouyet and Kimberly J. Gilbert | <p>A major goal of molecular evolutionary biology is to identify loci or regions of the genome under selection versus those evolving in a neutral manner. Correct identification allows accurate inference of the evolutionary process and thus compreh... | | Genome Evolution, Population Genetics / Genomics | Guillaume Achaz | 2019-09-26 10:58:10 | ||
29 Nov 2023

Does sociality affect evolutionary speed?On the evolutionary implications of being a social animalRecommended by Michael D Greenfield based on reviews by Rafael Lucas Rodriguez and 1 anonymous reviewerWhat does it mean to be highly social? Considering the so-called four ‘pinnacles’ of animal society (Wilson, 1975) – humans, cooperative breeding as found in some non-human mammals and birds, the social insects, and colonial marine invertebrates – having inter-individual relations extending beyond the sexual pair and the parent-offspring interaction is foremost. In many cases being social implies a high local population density, interaction with the same group of individuals over an extended time period, and an overlapping of generations. Additional features of social species may be a wide geographical range, perhaps associated with ecological and behavioral plasticity, the latter often facilitated by cultural transmission of traditions. Narrowing our perspective to the domain of PCI Evolutionary Biology, we might continue our question by asking whether being social predisposes one to a special evolutionary path toward the future. Do social species evolve faster (or slower) than their more solitary relatives such that over time they are more unlike (or similar to) those relatives (anagenesis)? And are evolutionary changes in social species more or less likely to be accompanied by lineage splitting (cladogenesis) and ultimately speciation? The latter question is parallel to one first posed over 40 years ago (West-Eberhard, 1979; Lande, 1981) for sexually selected traits: Do strong mating preferences and conspicuous courtship signals generate speciation via the Fisherian process or ecological divergence? An extensive survey of birds had found little supporting evidence (Price, 1998), but a recent one that focused on plumage complexity in tanagers did reveal a relationship, albeit a weak one (Price-Waldman et al., 2020). Because sexual selection has been viewed as a part of the broader process of social selection (West-Eberhard, 1979), it is thus fitting to extend our surveys to the evolutionary implications of being social. Unlike the inquiry for a sexual selection - evolutionary change connection, a social behavior counterpart has remained relatively untreated. Diverse logistical problems might account for this oversight. What objective proxies can be used for social behavior, and for the rate of evolutionary change within a lineage? How many empirical studies have generated data from which appropriate proxies could be extracted? More intractable is the conundrum arising from the connectedness between socially- and sexually-selected traits. For example, the elevated population density found in highly social species can greatly increase the mating advantage enjoyed by an attractive male. If anagenesis is detected, did it result from social behavior or sexual selection? And if social behavior leads to a group structure in which male-male competition is reduced, would a modest rate of evolutionary change be support for the sexual selection - evolutionary speed connection or evidence opposing the sociality - evolution one? Against the above odds, several biologists have begun to explore the notion that social behavior just might favor evolutionary speed in either anagenesis or cladogenesis. In a recent analysis relying on the comparative method, Lluís Socias-Martínez and Louise Rachel Peckre (2023) combed the scientific literature archives and identified those studies with specific data on the relationships between sexual selection or social behavior and evolutionary change, either anagenesis or cladogenesis. The authors were careful to employ fairly conservative criteria for including studies, and the number eventually retained was small. Nonetheless, some patterns emerge: Many more studies report anagenesis than cladogenesis, and many more report correlations with sexually-selected traits than with non-sexual social behavior ones. And, no study indicates a potential effect of social behavior on cladogenesis. Is this latter observation authentic or an artifact of a paucity of data? There are some a priori reasons why cladogenesis may seldom arise. Whereas highly social behavior could lead to fission encompassing mutually isolated population clusters within a species, social behavior may also engender counterbalancing plasticity that allows and even promotes inter-cluster migration and fusion. And briefly – and non-systematically, as the rate of lineage splitting would need to be measured – looking at one of the pinnacles of animal social behavior, the social insects, there is little indication that diversification has been accelerated. There are fewer than 3000 described species of termites, only ca. 16,000 ants, and the vast majority of bees and wasps are solitary. Lluís Socias-Martínez and Louise Rachel Peckre provide us with a very detailed discussion of these and a myriad of other complications. I end with a common refrain, we need more consideration of the authors’ interesting question, and much more data and analysis. One can thank Socias-Martínez and Peckre for pointing us in that direction. References Lande, R. (1981). Models of speciation by sexual selection on polygenic traits. Proc. Natn. Acad. Sci. USA 78, 3721-3725. https://doi.org/10.1073/pnas.78.6.3721 Price, T. (1998). Sexual selection and natural selection in bird speciation. Phil. Trans. Roy. Soc. B, 353, 251-260. https://doi.org/10.1098/rstb.1998.0207 Price‐Waldman, R. M., Shultz, A. J., & Burns, K. J. (2020). Speciation rates are correlated with changes in plumage color complexity in the largest family of songbirds. Evolution, 74(6), 1155–1169. https://doi.org/10.1111/evo.13982 Socias-Martínez and Peckre. (2023). Does sociality affect evolutionary speed? Zenodo, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.5281/zenodo.10086186 West-Eberhard, M. J. (1979). Sexual selection, social competition, and evolution. Proceedings of the American Philosophical Society, 123(4), 222–234. http://www.jstor.org/stable/2828804 Wilson, E. O. (1975). Sociobiology. The New Synthesis. Cambridge, Mass., The Belknap Press of Harvard University | Does sociality affect evolutionary speed? | Lluís Socias-Martínez, Louise Rachel Peckre | <p>An overlooked source of variation in evolvability resides in the social lives of animals. In trying to foster research in this direction, we offer a critical review of previous work on the link between evolutionary speed and sociality. A first ... | | Behavior & Social Evolution, Evolutionary Dynamics, Evolutionary Theory, Genome Evolution, Macroevolution, Molecular Evolution, Population Genetics / Genomics, Sexual Selection, Speciation | Michael D Greenfield | 2023-03-03 00:10:49 | ||
04 Nov 2020
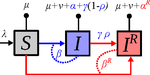
Treating symptomatic infections and the co-evolution of virulence and drug resistanceMore intense symptoms, more treatment, more drug-resistance: coevolution of virulence and drug-resistanceRecommended by Ludek Berec based on reviews by 3 anonymous reviewersMathematical models play an essential role in current evolutionary biology, and evolutionary epidemiology is not an exception [1]. While the issues of virulence evolution and drug-resistance evolution resonate in the literature for quite some time [2, 3], the study by Alizon [4] is one of a few that consider co-evolution of both these traits [5]. The idea behind this study is the following: treating individuals with more severe symptoms at a higher rate (which appears to be quite natural) leads to an appearance of virulent drug-resistant strains, via treatment failure. The author then shows that virulence in drug-resistant strains may face different selective pressures than in drug-sensitive strains and hence proceed at different rates. Hence, treatment itself modulates evolution of virulence. As one of the reviewers emphasizes, the present manuscript offers a mathematical view on why the resistant and more virulent strains can be selected in epidemics. Also, we both find important that the author highlights that the topic and results of this study can be attributed to public health policies and development of optimal treatment protocols [6]. References [1] Gandon S, Day T, Metcalf JE, Grenfell BT (2016) Forecasting epidemiological and evolutionary dynamics of infectious diseases. Trends Ecol Evol 31: 776-788. doi: https://doi.org/10.1016/j.tree.2016.07.010 | Treating symptomatic infections and the co-evolution of virulence and drug resistance | Samuel Alizon | <p>Antimicrobial therapeutic treatments are by definition applied after the onset of symptoms, which tend to correlate with infection severity. Using mathematical epidemiology models, I explore how this link affects the coevolutionary dynamics bet... | | Evolutionary Applications, Evolutionary Dynamics, Evolutionary Epidemiology, Evolutionary Theory | Ludek Berec | 2020-03-04 10:18:39 | ||
03 Jun 2019

Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory networkEarly and late flowering gene expression patterns in maizeRecommended by Tanja Pyhäjärvi based on reviews by Laura Shannon and 2 anonymous reviewersArtificial selection experiments are key experiments in evolutionary biology. The demonstration that application of selective pressure across multiple generations results in heritable phenotypic changes is a tangible and reproducible proof of the evolution by natural selection. References [1] Hill, W. G., & Caballero, A. (1992). Artificial selection experiments. Annual Review of Ecology and Systematics, 23(1), 287-310. doi: 10.1146/annurev.es.23.110192.001443 | Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network | Maud Irène Tenaillon, Khawla Sedikki, Maeva Mollion, Martine Le Guilloux, Elodie Marchadier, Adrienne Ressayre, Christine Dillmann | <p>Artificial selection experiments are designed to investigate phenotypic evolution of complex traits and its genetic basis. Here we focused on flowering time, a trait of key importance for plant adaptation and life-cycle shifts. We undertook div... | | Adaptation, Experimental Evolution, Expression Studies, Quantitative Genetics | Tanja Pyhäjärvi | 2018-11-23 11:57:35 | ||
18 May 2018

Modularity of genes involved in local adaptation to climate despite physical linkageDifferential effect of genes in diverse environments, their role in local adaptation and the interference between genes that are physically linkedRecommended by Sebastian Ernesto Ramos-Onsins based on reviews by Tanja Pyhäjärvi and 1 anonymous reviewerThe genome of eukaryotic species is a complex structure that experience many different interactions within itself and with the surrounding environment. The genetic architecture of a phenotype (that is, the set of genetic elements affecting a trait of the organism) plays a fundamental role in understanding the adaptation process of a species to, for example, different climate environments, or to its interaction with other species. Thus, it is fundamental to study the different aspects of the genetic architecture of the species and its relationship with its surronding environment. Aspects such as modularity (the number of genetic units and the degree to which each unit is affecting a trait of the organism), pleiotropy (the number of different effects that a genetic unit can have on an organism) or linkage (the degree of association between the different genetic units) are essential to understand the genetic architecture and to interpret the effects of selection on the genome. Indeed, the knowledge of the different aspects of the genetic architecture could clarify whether genes are affected by multiple aspects of the environment or, on the contrary, are affected by only specific aspects [1,2]. The work performed by Lotterhos et al. [3] sought to understand the genetic architecture of the adaptation to different environments in lodgepole pine (Pinus contorta), considering as candidate SNPs those previously detected as a result of its extreme association patterns to different environmental variables or to extreme population differentiation. This consideration is very important because the study is only relevant if the studied markers are under the effect of selection. Otherwise, the genetic architecture of the adaptation to different environments would be masked by other (neutral) kind of associations that would be difficult to interpret [4,5]. In order to understand the relationship between genetic architecture and adaptation, it is relevant to detect the association networks of the candidate SNPs with climate variables (a way to measure modularity) and if these SNPs (and loci) are affected by single or multiple environments (a way to measure pleiotropy). The authors used co-association networks, an innovative approach in this field, to analyse the interaction between the environmental information and the genetic polymorphism of each individual. This methodology is more appropriate than other multivariate methods - such as analysis based on principal components - because it is possible to cluster SNPs based on associations with similar environmental variables. In this sense, the co-association networks allowed to both study the genetic and physical linkage between different co-associations modules but also to compare two different models of evolution: a Modular environmental response architecture (specific genes are affected by specific aspects of the environment) or a Universal pleiotropic environmental response architecture (all genes are affected by all aspects of the environment). The representation of different correlations between allelic frequency and environmental factors (named galaxy biplots) are especially informative to understand the effect of the different clusters on specific aspects of the environment (for example, the co-association network ‘Aridity’ shows strong associations with hot/wet versus cold/dry environments). The analysis performed by Lotterhos et al. [3], although it has some unavoidable limitations (e.g., only extreme candidate SNPs are selected, limiting the results to the stronger effects; the genetic and physical map is incomplete in this species), includes relevant results and also implements new methodologies in the field. To highlight some of them: the preponderance of a Modular environmental response architecture (evolution in separated modules), the detection of physical linkage among SNPs that are co-associated with different aspects of the environment (which was unexpected a priori), the implementation of co-association networks and galaxy biplots to see the effect of modularity and pleiotropy on different aspects of environment. Finally, this work contains remarkable introductory Figures and Tables explaining unambiguously the main concepts [6] included in this study. This work can be treated as a starting point for many other future studies in the field. References [1] Hancock AM, Brachi B, Faure N, Horton MW, Jarymowycz LB, Sperone FG, Toomajian C, Roux F & Bergelson J. 2011. Adaptation to climate across the Arabidopsis thaliana genome. Science 334: 83–86. doi: 10.1126/science.1209244 | Modularity of genes involved in local adaptation to climate despite physical linkage | Katie E. Lotterhos, Sam Yeaman, Jon Degner, Sally Aitken, Kathryn Hodgins | <p>Background: Physical linkage among genes shaped by different sources of selection is a fundamental aspect of genetic architecture. Theory predicts that evolution in complex environments selects for modular genetic architectures and high recombi... | | Adaptation, Bioinformatics & Computational Biology, Genome Evolution | Sebastian Ernesto Ramos-Onsins | 2017-10-15 19:21:57 | ||
04 Mar 2024
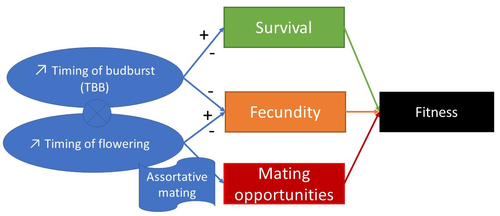
Interplay between fecundity, sexual and growth selection on the spring phenology of European beech (Fagus sylvatica L.).Interplay between fecundity, sexual and growth selection on the spring phenology of European beech (Fagus sylvatica L.)Recommended by Santiago C. Gonzalez-Martinez based on reviews by 2 anonymous reviewers
Starting with the seminar paper by Lande & Arnold (1983), several studies have addressed phenotypic selection in natural populations of a wide variety of organisms, with a recent renewed interest in forest trees (e.g., Oddou-Muratorio et al. 2018; Alexandre et al. 2020; Westergren et al. 2023). Because of their long generation times, long-lived organisms such as forest trees may suffer the most from maladaptation due to climate change, and whether they will be able to adapt to new environmental conditions in just one or a few generations is hotly debated. In this study, Oddou-Muratorio and colleagues (2024) extend the current framework to add two additional selection components that may alter patterns of fecundity selection and the estimation of standard selection gradients, namely sexual selection (evaluated as differences in flowering phenology conducting to assortative mating) and growth (viability) selection. Notably, the study is conducted in two contrasted environments (low vs high altitude populations) providing information on how the environment may modulate selection patterns in spring phenology. Spring phenology is a key adaptive trait that has been shown to be already affected by climate change in forest trees (Alberto et al. 2013). While fecundity selection for early phenology has been extensively reported before (see Munguía-Rosas et al. 2011), the authors found that this kind of selection can be strongly modulated by sexual selection, depending on the environment. Moreover, they found a significant correlation between early phenology and seedling growth in a common garden, highlighting the importance of this trait for early survival in European beech. As a conclusion, this original research puts in evidence the need for more integrative approaches for the study of natural selection in the field, as well as the importance of testing multiple environments and the relevance of common gardens to further evaluate phenotypic changes due to real-time selection. PS: The recommender and the first author of the preprint have shared authorship in a recent paper in a similar topic (Westergren et al. 2023). Nevertheless, the recommender has not contributed in any way or was aware of the content of the current preprint before acting as recommender, and steps have been taken for a fair and unpartial evaluation. References Alberto, F. J., Aitken, S. N., Alía, R., González‐Martínez, S. C., Hänninen, H., Kremer, A., Lefèvre, F., Lenormand, T., Yeaman, S., Whetten, R., & Savolainen, O. (2013). Potential for evolutionary responses to climate change - evidence from tree populations. Global Change Biology, 19(6), 1645‑1661. Oddou-Muratorio S, Bontemps A, Gauzere J, Klein E (2024) Interplay between fecundity, sexual and growth selection on the spring phenology of European beech (Fagus sylvatica L.). bioRxiv, 2023.04.27.538521, ver. 2 peer-reviewed and recommended by Peer Community In Evolutionary Biology https://doi.org/10.1101/2023.04.27.538521 Oddou-Muratorio, S., Gauzere, J., Bontemps, A., Rey, J.-F., & Klein, E. K. (2018). Tree, sex and size: Ecological determinants of male vs. female fecundity in three Fagus sylvatica stands. Molecular Ecology, 27(15), 3131‑3145. | Interplay between fecundity, sexual and growth selection on the spring phenology of European beech (*Fagus sylvatica* L.). | Sylvie Oddou-Muratorio, Aurore Bontemps, Julie Gauzere, Etienne Klein | <p>Background: Plant phenological traits such as the timing of budburst or flowering can evolve on ecological timescales through response to fecundity and viability selection. However, interference with sexual selection may arise from assortative ... | | Adaptation, Evolutionary Ecology, Quantitative Genetics, Reproduction and Sex, Sexual Selection | Santiago C. Gonzalez-Martinez | 2023-05-02 11:57:23 | ||
28 Mar 2019
Ancient tropical extinctions contributed to the latitudinal diversity gradientOne (more) step towards a dynamic view of the Latitudinal Diversity GradientRecommended by Joaquín Hortal and Juan Arroyo based on reviews by Juan Arroyo, Joaquín Hortal, Arne Mooers, Joaquin Calatayud and 2 anonymous reviewers
The Latitudinal Diversity Gradient (LDG) has fascinated natural historians, ecologists and evolutionary biologists ever since [1] described it about 200 years ago [2]. Despite such interest, agreement on the origin and nature of this gradient has been elusive. Several tens of hypotheses and models have been put forward as explanations for the LDG [2-3], that can be grouped in ecological, evolutionary and historical explanations [4] (see also [5]). These explanations can be reduced to no less than 26 hypotheses, which account for variations in ecological limits for the establishment of progressively larger assemblages, diversification rates, and time for species accumulation [5]. Besides that, although in general the tropics hold more species, different taxa show different shapes and rates of spatial variation [6], and a considerable number of groups show reverse patterns, with richer assemblages in cold temperate regions (see e.g. [7-9]). References | Ancient tropical extinctions contributed to the latitudinal diversity gradient | Andrea S. Meseguer, Fabien Condamine | <p>Biodiversity currently peaks at the equator, decreasing toward the poles. Growing fossil evidence suggest that this hump-shaped latitudinal diversity gradient (LDG) has not been persistent through time, with similar species diversity across lat... | | Evolutionary Dynamics, Evolutionary Ecology, Macroevolution, Paleontology, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Joaquín Hortal | 2017-12-20 14:58:01 | ||
24 Aug 2022

Density dependent environments can select for extremes of body sizeA population biological modeling approach for life history and body size evolutionRecommended by Wolf Blanckenhorn based on reviews by Frédéric Guillaume and 2 anonymous reviewersBody size evolution is a central theme in evolutionary biology. Particularly the question of when and how smaller body sizes can evolve continues to interest evolutionary ecologists, because most life history models, and the empirical evidence, document that large body size is favoured by natural and sexual selection in most (even small) organisms and environments at most times. How, then, can such a large range of body size and life history syndromes evolve and coexist in nature? The paper by Coulson et al. lifts this question to the level of the population, a relatively novel approach using so-called integral projection (simulation) models (IPMs) (as opposed to individual-based or game theoretical models). As is well outlined by (anonymous) Reviewer 1, and following earlier papers spearheading this approach in other life history contexts, the authors use the well-known carrying capacity (K) of population biology as the ultimate fitness parameter to be maximized or optimized (rather than body size per se), to ultimately identify factors and conditions promoting the evolution of extreme body sizes in nature. They vary (individual or population) size-structured growth trajectories to observe age and size at maturity, surivorship and fecundity/fertility schedules upon evaluating K (see their Fig. 1). Importantly, trade-offs are introduced via density-dependence, either for adult reproduction or for juvenile survival, in two (of several conceivable) basic scenarios (see their Table 2). All other relevant standard life history variables (see their Table 1) are assumed density-independent, held constant or zero (as e.g. the heritability of body size). The authors obtain evidence for disruptive selection on body size in both scenarios, with small size and a fast life history evolving below a threshold size at maturity (at the lowest K) and large size and a slow life history beyond this threshold (see their Fig. 2). Which strategy wins ultimately depends on the fitness benefits of delaying sexual maturity (at larger size and longer lifespan) at the adult stage relative to the preceeding juvenile mortality costs, in agreement with classic life history theory (Roff 1992, Stearns 1992). The modeling approach can be altered and refined to be applied to other key life history parameters and environments. These results can ultimately explain the evolution of smaller body sizes from large body sizes, or vice versa, and their corresponding life history syndromes, depending on the precise environmental circumstances. All reviewers agreed that the approach taken is technically sound (as far as it could be evaluated), and that the results are interesting and worthy of publication. In a first round of reviews various clarifications of the manuscript were suggested by the reviewers. The new version was substantially changed by the authors in response, to the extent that it now is a quite different but much clearer paper with a clear message palatable for the general reader. The writing is now to the point, the paper's focus becomes clear in the Introduction, Methods & Results are much less technical, the Figures illustrative, and the descriptions and interpretations in the Discussion are easy to follow. In general any reader may of course question the choice and realism of the scenarios and underlying assumptions chosen by the authors for simplicity and clarity, for instance no heritability of body size and no cost of reproduction (other than mortality). But this is always the case in modeling work, and the authors acknowledge and in fact suggest concrete extensions and expansions of their approach in the Discussion. References Coulson T., Felmy A., Potter T., Passoni G., Montgomery R.A., Gaillard J.-M., Hudson P.J., Travis J., Bassar R.D., Tuljapurkar S., Marshall D.J., Clegg S.M. (2022) Density-dependent environments can select for extremes of body size. bioRxiv, 2022.02.17.480952, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.02.17.480952 | Density dependent environments can select for extremes of body size | Tim Coulson, Anja Felmy, Tomos Potter, Gioele Passoni, Robert A Montgomery, Jean-Michel Gaillard, Peter J Hudson, Joseph Travis, Ronald D Bassar, Shripad D Tuljapurkar, Dustin Marshall, Sonya M Clegg | <p>Body size variation is an enigma. We do not understand why species achieve the sizes they do, and this means we also do not understand the circumstances under which gigantism or dwarfism is selected. We develop size-structured integral projecti... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Theory, Life History | Wolf Blanckenhorn | 2022-02-21 07:59:04 |




