
Latest recommendations

| Id | Title | Authors▲ | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
22 Sep 2020
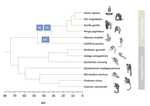
Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primatesStudying genetic antagonisms as drivers of genome evolutionRecommended by Mathieu Joron based on reviews by Qi Zhou and 3 anonymous reviewersSex chromosomes are special in the genome because they are often highly differentiated over much of their lengths and marked by degenerative evolution of their gene content. Understanding why sex chromosomes differentiate requires deciphering the forces driving their recombination patterns. Suppression of recombination may be subject to selection, notably because of functional effects of locking together variation at different traits, as well as longer-term consequences of the inefficient purge of deleterious mutations, both of which may contribute to patterns of differentiation [1]. As an example, male and female functions may reveal intrinsic antagonisms over the optimal genotypes at certain genes or certain combinations of interacting genes. As a result, selection may favour the recruitment of rearrangements blocking recombination and maintaining the association of sex-antagonistic allele combinations with the sex-determining locus. References [1] Charlesworth D (2017) Evolution of recombination rates between sex chromosomes. Philosophical Transactions of the Royal Society B: Biological Sciences, 372, 20160456. https://doi.org/10.1098/rstb.2016.0456 | Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates | Rylan Shearn, Alison E. Wright, Sylvain Mousset, Corinne Régis, Simon Penel, Jean-François Lemaitre, Guillaume Douay, Brigitte Crouau-Roy, Emilie Lecompte, Gabriel A.B. Marais | <p>Sex chromosomes are typically comprised of a non-recombining region and a recombining pseudoautosomal region. Accurately quantifying the relative size of these regions is critical for sex chromosome biology both from a functional (i.e. number o... | | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Reproduction and Sex, Sexual Selection | Mathieu Joron | 2019-02-04 15:16:32 | ||
12 Jul 2017

Despite reproductive interference, the net outcome of reproductive interactions among spider mite species is not necessarily costlyThe pros and cons of mating with strangersRecommended by Vincent Calcagno based on reviews by Joël Meunier and Michael D Greenfield
Interspecific matings are by definition rare events in nature, but when they occur they can be very important, and not only because they might condition gene flow between species. Even when such matings have no genetic consequence, for instance if they do not yield any fertile hybrid offspring, they can still have an impact on the population dynamics of the species involved [1]. Such atypical pairings between heterospecific partners are usually regarded as detrimental or undesired; as they interfere with the occurrence or success of intraspecific matings, they are expected to cause a decline in absolute fitness. References [1] Gröning J. & Hochkirch A. 2008. Reproductive interference between animal species. The Quarterly Review of Biology 83: 257-282. doi: 10.1086/590510 [2] Clemente SH, Santos I, Ponce AR, Rodrigues LR, Varela SAM & Magalhaes S. 2017 Despite reproductive interference, the net outcome of reproductive interactions among spider mite species is not necessarily costly. bioRxiv 113274, ver. 4 of the 30th of June 2017. doi: 10.1101/113274 | Despite reproductive interference, the net outcome of reproductive interactions among spider mite species is not necessarily costly | Salomé H. Clemente, Inês Santos, Rita Ponce, Leonor R. Rodrigues, Susana A. M. Varela and Sara Magalhães | Reproductive interference is considered a strong ecological force, potentially leading to species exclusion. This supposes that the net effect of reproductive interactions is strongly negative for one of the species involved. Testing this requires... | | Behavior & Social Evolution, Evolutionary Ecology, Species interactions | Vincent Calcagno | 2017-03-06 11:48:08 | ||
04 Nov 2020
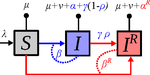
Treating symptomatic infections and the co-evolution of virulence and drug resistanceMore intense symptoms, more treatment, more drug-resistance: coevolution of virulence and drug-resistanceRecommended by Ludek Berec based on reviews by 3 anonymous reviewersMathematical models play an essential role in current evolutionary biology, and evolutionary epidemiology is not an exception [1]. While the issues of virulence evolution and drug-resistance evolution resonate in the literature for quite some time [2, 3], the study by Alizon [4] is one of a few that consider co-evolution of both these traits [5]. The idea behind this study is the following: treating individuals with more severe symptoms at a higher rate (which appears to be quite natural) leads to an appearance of virulent drug-resistant strains, via treatment failure. The author then shows that virulence in drug-resistant strains may face different selective pressures than in drug-sensitive strains and hence proceed at different rates. Hence, treatment itself modulates evolution of virulence. As one of the reviewers emphasizes, the present manuscript offers a mathematical view on why the resistant and more virulent strains can be selected in epidemics. Also, we both find important that the author highlights that the topic and results of this study can be attributed to public health policies and development of optimal treatment protocols [6]. References [1] Gandon S, Day T, Metcalf JE, Grenfell BT (2016) Forecasting epidemiological and evolutionary dynamics of infectious diseases. Trends Ecol Evol 31: 776-788. doi: https://doi.org/10.1016/j.tree.2016.07.010 | Treating symptomatic infections and the co-evolution of virulence and drug resistance | Samuel Alizon | <p>Antimicrobial therapeutic treatments are by definition applied after the onset of symptoms, which tend to correlate with infection severity. Using mathematical epidemiology models, I explore how this link affects the coevolutionary dynamics bet... | | Evolutionary Applications, Evolutionary Dynamics, Evolutionary Epidemiology, Evolutionary Theory | Ludek Berec | 2020-03-04 10:18:39 | ||
14 May 2020

Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisonsFrom populations to subspecies… to species? Contrasting patterns of local adaptation in closely-related taxa and their potential contribution to species divergenceRecommended by Emmanuelle Porcher based on reviews by Sophie Karrenberg, Santiago C. Gonzalez-Martinez and 1 anonymous reviewerElevation gradients are convenient and widely used natural setups to study local adaptation, particularly in these times of rapid climate change [e.g. 1]. Marin and her collaborators [2] did not follow the mainstream, however. Instead of tackling adaptation to climate change, they used elevation gradients to address another crucial evolutionary question [3]: could adaptation to altitude lead to ecological speciation, i.e. reproductive isolation between populations in spite of gene flow? More specifically, they examined how much local adaptation to environmental variation differed among closely-related, recently diverged subspecies. They studied several populations of two subspecies of snapdragon (Antirrhinum majus), with adjacent geographical distributions. Using common garden experiments and the classical, but still useful, QST-FST comparison, they demonstrate contrasting patterns of local adaptation to altitude between the two subspecies, with several traits under divergent selection in A. majus striatum but none in A. majus pseudomajus. These differences in local adaptation may contribute to species divergence, and open many stimulating questions on the underlying mechanisms, such as the identity of environmental drivers or contribution of reproductive isolation involving flower color polymorphism. References [1] Anderson, J. T., and Wadgymar, S. M. (2020). Climate change disrupts local adaptation and favours upslope migration. Ecology letters, 23(1), 181-192. doi: 10.1111/ele.13427 | Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisons | Sara Marin, Anaïs Gibert, Juliette Archambeau, Vincent Bonhomme, Mylène Lascoste and Benoit Pujol | <p>Phenotypic divergence among natural populations can be explained by natural selection or by neutral processes such as drift. Many examples in the literature compare putatively neutral (FST) and quantitative genetic (QST) differentiation in mult... | | Adaptation, Evolutionary Ecology, Genotype-Phenotype, Morphological Evolution, Quantitative Genetics | Emmanuelle Porcher | 2018-08-05 15:34:30 | ||
24 Jan 2017

POSTPRINT
Birth of a W sex chromosome by horizontal transfer of Wolbachia bacterial symbiont genomeA newly evolved W(olbachia) sex chromosome in pillbug!Recommended by Gabriel Marais and Sylvain CharlatIn some taxa such as fish and arthropods, closely related species can have different mechanisms of sex determination and in particular different sex chromosomes, which implies that new sex chromosomes are constantly evolving [1]. Several models have been developed to explain this pattern but empirical data are lacking and the causes of the fast sex chromosome turn over remain mysterious [2-4]. Leclerq et al. [5] in a paper that just came out in PNAS have focused on one possible explanation: Wolbachia. This widespread intracellular symbiont of arthropods can manipulate its host reproduction in a number of ways, often by biasing the allocation of resources toward females, the transmitting sex. Perhaps the most spectacular example is seen in pillbugs, where Wolbachia commonly turns infected males into females, thus doubling its effective transmission to grandchildren. Extensive investigations on this phenomenon were initiated 30 years ago in the host species Armadillidium vulgare. The recent paper by Leclerq et al. beautifully validates an hypothesis formulated in these pioneer studies [6], namely, that a nuclear insertion of the Wolbachia genome caused the emergence of new female determining chromosome, that is, a new sex chromosome. Many populations of A. vulgare are infected by the feminising Wolbachia strain wVulC, where the spread of the bacterium has also induced the loss of the ancestral female determining W chromosome (because feminized ZZ individuals produce females without transmitting any W). In these populations, all individuals carry two Z chromosomes, so that the bacterium is effectively the new sex-determining factor: specimens that received Wolbachia from their mother become females, while the occasional loss of Wolbachia from mothers to eggs allows the production of males. Intriguingly, studies from natural populations also report that some females are devoid both of Wolbachia and the ancestral W chromosome, suggesting the existence of new female determining nuclear factor, the hypothetical “f element”. Leclerq et al. [5] found the f element and decrypted its origin. By sequencing the genome of a strain carrying the putative f element, they found that a nearly complete wVulC genome got inserted in the nuclear genome and that the chromosome carrying the insertion has effectively become a new W chromosome. The insertion is indeed found only in females, PCRs and pedigree analysis tell. Although the Wolbachia-derived gene(s) that became sex-determining gene(s) remain to be identified among many possible candidates, the genomic and genetic evidence are clear that this Wolbachia insertion is determining sex in this pillbug strain. Leclerq et al. [5] also found that although this insertion is quite recent, many structural changes (rearrangements, duplications) have occurred compared to the wVulC genome, which study will probably help understand which bacterial gene(s) have retained a function in the nucleus of the pillbug. Also, in the future, it will be interesting to understand how and why exactly the nuclear inserted Wolbachia rose in frequency in the pillbug population and how the cytoplasmic Wolbachia was lost, and to tease apart the roles of selection and drift in this event. We highly recommend this paper, which provides clear evidence that Wolbachia has caused sex chromosome turn over in one species, opening the conjecture that it might have done so in many others. References [1] Bachtrog D, Mank JE, Peichel CL, Kirkpatrick M, Otto SP, Ashman TL, Hahn MW, Kitano J, Mayrose I, Ming R, Perrin N, Ross L, Valenzuela N, Vamosi JC. 2014. Tree of Sex Consortium. Sex determination: why so many ways of doing it? PLoS Biology 12: e1001899. doi: 10.1371/journal.pbio.1001899 [2] van Doorn GS, Kirkpatrick M. 2007. Turnover of sex chromosomes induced by sexual conflict. Nature 449: 909-912. doi: 10.1038/nature06178 [3] Cordaux R, Bouchon D, Grève P. 2011. The impact of endosymbionts on the evolution of host sex-determination mechanisms. Trends in Genetics 27: 332-341. doi: 10.1016/j.tig.2011.05.002 [4] Blaser O, Neuenschwander S, Perrin N. 2014. Sex-chromosome turnovers: the hot-potato model. American Naturalist 183: 140-146. doi: 10.1086/674026 [5] Leclercq S, Thézé J, Chebbi MA, Giraud I, Moumen B, Ernenwein L, Grève P, Gilbert C, Cordaux R. 2016. Birth of a W sex chromosome by horizontal transfer of Wolbachia bacterial symbiont genome. Proceeding of the National Academy of Science USA 113: 15036-15041. doi: 10.1073/pnas.1608979113 [6] Legrand JJ, Juchault P. 1984. Nouvelles données sur le déterminisme génétique et épigénétique de la monogénie chez le crustacé isopode terrestre Armadillidium vulgare Latr. Génétique Sélection Evolution 16: 57–84. doi: 10.1186/1297-9686-16-1-57 | Birth of a W sex chromosome by horizontal transfer of Wolbachia bacterial symbiont genome | Sébastien Leclercq, Julien Thézé, Mohamed Amine Chebbi, Isabelle Giraud, Bouziane Moumen, Lise Ernenwein, Pierre Grève, Clément Gilbert, and Richard Cordaux | Sex determination is an evolutionarily ancient, key developmental pathway governing sexual differentiation in animals. Sex determination systems are remarkably variable between species or groups of species, however, and the evolutionary forces und... | | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Reproduction and Sex, Species interactions | Gabriel Marais | 2017-01-13 15:15:51 | ||
15 Dec 2016

POSTPRINT
Limiting opportunities for cheating stabilizes virulence in insect parasitic nematodesApplication of kin theory to long-standing problem in nematode production for biocontrolRecommended by Thomas Sappington and Ruth Arabelle HufbauerMuch research effort has been extended toward developing systems for managing soil inhabiting insect pests of crops with entomopathogenic nematodes as biocontrol agents. Although small plot or laboratory experiments may suggest a particular insect pest is vulnerable to management in this way, it is often difficult to scale-up nematode production for application at the field- and farm scale to make such a tactic viable. Part of the problem is that entomopathogenic nematode strains must be propagated by serial passage in vivo, because storage by freezing decreases fitness. At the same time, serial propagation results in loss of virulence (ability to infect) over generations in the laboratory, a phenomenon called attenuation. To probe the underlying reasons for development of attenuation, as a prerequisite to designing strategies to mitigate it, Shapiro-Ilan and Raymond [1] turned to evolutionary theory of social conflict as a possible explanatory framework. Virulence of entomopathogenic nematodes depends on a combination of virulence factors, like various proteases, secreted by both the nematode and symbiotic bacteria to overcome host defenses. Attenuation is characterized in part by a reduced production of these factors. Invasion of a host involves simultaneous attack by a group of nematodes ("cooperators"), which together neutralize host defenses enough to allow individuals to successfully invade. "Cheaters" in the invading population can avoid the metabolic costs of producing virulence factors while reaping the benefits of infecting the host made vulnerable by the cooperators in the population. The authors hypothesize that an increase in frequency of cheaters may contribute to attenuation of virulence during serial propagation in the laboratory. The evolutionary dynamics of cheater frequency in a population have been explored in many contexts as part of kin selection theory. Cheaters can increase in a population by outcompeting cooperators in a host if overall relatedness within the invading population is low. Conversely, frequency of altruism, or costly cooperation, increases in a population if relatedness is high, which is enhanced by low effective dispersal. However, a population that is too isolated can suffer from inbreeding effects, and competition will occur mainly among relatives, which decreases the fitness benefits of altruism. Shapiro-Ilan and Raymond [1] tested changes in virulence and reproductive output in a serially propagated entomopathogenic nematode, Heterorhabditis floridensis. They compared lines of high or low relatedness, manipulated via multiplicity of infection (MOI) rates (where a low dose of nematodes gives high relatedness and a high dose gives low relatedness); and under global or local competition, manipulated by pooling populations emerging from all or only two host cadavers per generation, respectively. As predicted, treatments of high relatedness (low MOI) and global competition had the greatest level of reproduction, while all lines of low relatedness (high MOI) evolved decreased reproduction and decreased virulence, which led to extinction. The key finding was that lines in the high relatedness (low MOI) and low (local) competition treatment exhibited the most stable virulence through the 12 generations tested. Thus, to minimize attenuation of virulence while maintaining fitness of recently isolated entomopathogenic nematodes, the authors recommend insect hosts be inoculated with low doses of nematodes from inocula pools from as few cadavers as possible. The application of evolutionary theory, with a clever experimental design, to an important problem in pest management makes this paper particularly noteworthy. Reference [1] Shapiro-Ilan D, Raymond B. 2016. Limiting opportunities for cheating stabilizes virulence in insect parasitic nematodes. Evolutionary Applications 9:462-470. doi: 10.1111/eva.12348 | Limiting opportunities for cheating stabilizes virulence in insect parasitic nematodes | Shapiro-Ilan D. and B. Raymond | Cooperative secretion of virulence factors by pathogens can lead to social conflict when cheating mutants exploit collective secretion, but do not contribute to it. If cheats outcompete cooperators within hosts, this can cause loss of virulence.... | | Adaptation, Behavior & Social Evolution, Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory, Experimental Evolution, Population Genetics / Genomics, Reproduction and Sex | Thomas Sappington | 2016-12-15 18:33:39 | ||
09 Feb 2018
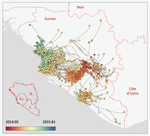
Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreakSimulating the effect of public health interventions using dated virus sequences and geographical dataRecommended by Samuel Alizon based on reviews by Christian Althaus, Chris Wymant and 1 anonymous reviewer based on reviews by Christian Althaus, Chris Wymant and 1 anonymous reviewer
Perhaps because of its deadliness, the 2013-2016 Ebola Virus (EBOV) epidemics in West-Africa has led to unprecedented publication and sharing of full virus genome sequences. This was both rapid (90 full genomes were shared within weeks [1]) and important (more than 1500 full genomes have been released overall [2]). Furthermore, the availability of the metadata (especially GPS location) has led to depth analyses of the geographical spread of the epidemics [3]. References [1] Gire et al. 2014. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345: 1369–1372. doi: 10.1126/science.1259657. | Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreak | Simon Dellicour, Guy Baele, Gytis Dudas, Nuno R. Faria, Oliver G. Pybus, Marc A. Suchard, Andrew Rambaut, Philippe Lemey | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (https://doi.org/10.24072/pci.evolbiol.100046). The recent Ebola virus (EBOV) outbreak in West Africa witnessed considerable efforts to obtain viral genom... | | Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Samuel Alizon | 2017-09-30 13:49:57 | ||
04 Sep 2019

The discernible and hidden effects of clonality on the genotypic and genetic states of populations: improving our estimation of clonal ratesHow to estimate clonality from genetic data: use large samples and consider the biology of the speciesRecommended by Myriam Heuertz based on reviews by David Macaya-Sanz, Marcela Van Loo and 1 anonymous reviewer
Population geneticists frequently use the genetic and genotypic information of a population sample of individuals to make inferences on the reproductive system of a species. The detection of clones, i.e. individuals with the same genotype, can give information on whether there is clonal (vegetative) reproduction in the species. If clonality is detected, population geneticists typically use genotypic richness R, the number of distinct genotypes relative to the sample size, to estimate the rate of clonality c, which can be defined as the proportion of reproductive events that are clonal. Estimating the rate of clonality based on genotypic richness is however problematic because, to date, there is no analytical, nor simulation-based, characterization of this relationship. Furthermore, the effect of sampling on this relationship has never been critically examined. References [1] Stoeckel, S., Porro, B., and Arnaud-Haond, S. (2019). The discernible and hidden effects of clonality on the genotypic and genetic states of populations: improving our estimation of clonal rates. ArXiv:1902.09365 [q-Bio] v4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. Retrieved from http://arxiv.org/abs/1902.09365v4 | The discernible and hidden effects of clonality on the genotypic and genetic states of populations: improving our estimation of clonal rates | Solenn Stoeckel, Barbara Porro, Sophie Arnaud-Haond | <p>Partial clonality is widespread across the tree of life, but most population genetics models are conceived for exclusively clonal or sexual organisms. This gap hampers our understanding of the influence of clonality on evolutionary trajectories... | | Population Genetics / Genomics, Reproduction and Sex | Myriam Heuertz | 2019-02-28 10:10:56 | ||
25 Feb 2021

Alteration of gut microbiota with a broad-spectrum antibiotic does not impair maternal care in the European earwigAssessing the role of host-symbiont interactions in maternal care behaviourRecommended by Trine Bilde based on reviews by Nadia Aubin-Horth, Gabrielle Davidson and 1 anonymous reviewer
The role of microbial symbionts in governing social traits of their hosts is an exciting and developing research area. Just like symbionts influence host reproductive behaviour and can cause mating incompatibilities to promote symbiont transmission through host populations (Engelstadter and Hurst 2009; Correa and Ballard 2016; Johnson and Foster 2018) (see also discussion on conflict resolution in Johnsen and Foster 2018), microbial symbionts could enhance transmission by promoting the social behaviour of their hosts (Ezenwa et al. 2012; Lewin-Epstein et al. 2017; Gurevich et al. 2020). Here I apply the term ‘symbiosis’ in the broad sense, following De Bary 1879 as “the living together of two differently named organisms“ independent of effects on the organisms involved (De Bary 1879), i.e. the biological interaction between the host and its symbionts may include mutualism, parasitism and commensalism. References Correa, C. C., and Ballard, J. W. O. (2016). Wolbachia associations with insects: winning or losing against a master manipulator. Frontiers in Ecology and Evolution, 3, 153. doi: https://doi.org/10.3389/fevo.2015.00153 De Bary, A. (1879). Die Erscheinung der Symbiose. Verlag von Karl J. Trubner, Strassburg. Engelstädter, J., and Hurst, G. D. (2009). The ecology and evolution of microbes that manipulate host reproduction. Annual Review of Ecology, Evolution, and Systematics, 40, 127-149. doi: https://doi.org/10.1146/annurev.ecolsys.110308.120206 Ezenwa, V. O., Gerardo, N. M., Inouye, D. W., Medina, M., and Xavier, J. B. (2012). Animal behavior and the microbiome. Science, 338(6104), 198-199. doi: https://doi.org/10.1126/science.1227412 Gurevich, Y., Lewin-Epstein, O., and Hadany, L. (2020). The evolution of paternal care: a role for microbes?. Philosophical Transactions of the Royal Society B, 375(1808), 20190599. doi: https://doi.org/10.1098/rstb.2019.0599 Hird, S. M. (2017). Evolutionary biology needs wild microbiomes. Frontiers in microbiology, 8, 725. doi: https://doi.org/10.3389/fmicb.2017.00725 Johnson, K. V. A., and Foster, K. R. (2018). Why does the microbiome affect behaviour?. Nature reviews microbiology, 16(10), 647-655. doi: https://doi.org/10.1038/s41579-018-0014-3 Kramer et al. (2017). When earwig mothers do not care to share: parent–offspring competition and the evolution of family life. Functional Ecology, 31(11), 2098-2107. doi: https://doi.org/10.1111/1365-2435.12915 Lewin-Epstein, O., Aharonov, R., and Hadany, L. (2017). Microbes can help explain the evolution of host altruism. Nature communications, 8(1), 1-7. doi: https://doi.org/10.1038/ncomms14040 Meunier, J., and Kölliker, M. (2012). Parental antagonism and parent–offspring co-adaptation interact to shape family life. Proceedings of the Royal Society B: Biological Sciences, 279(1744), 3981-3988. doi: https://doi.org/10.1098/rspb.2012.1416 Meunier, J., Wong, J. W., Gómez, Y., Kuttler, S., Röllin, L., Stucki, D., and Kölliker, M. (2012). One clutch or two clutches? Fitness correlates of coexisting alternative female life-histories in the European earwig. Evolutionary Ecology, 26(3), 669-682. doi: https://doi.org/10.1007/s10682-011-9510-x Nalepa, C. A. (2020). Origin of mutualism between termites and flagellated gut protists: transition from horizontal to vertical transmission. Frontiers in Ecology and Evolution, 8, 14. doi: https://doi.org/10.3389/fevo.2020.00014 Ratz, T., Kramer, J., Veuille, M., and Meunier, J. (2016). The population determines whether and how life-history traits vary between reproductive events in an insect with maternal care. Oecologia, 182(2), 443-452. doi: https://doi.org/10.1007/s00442-016-3685-3 Van Meyel, S., Devers, S., Dupont, S., Dedeine, F. and Meunier, J. (2021) Alteration of gut microbiota with a broad-spectrum antibiotic does not impair maternal care in the European earwig. bioRxiv, 2020.10.08.331363. ver. 5 peer-reviewed and recommended by PCI Evol Biol. https://doi.org/10.1101/2020.10.08.331363 | Alteration of gut microbiota with a broad-spectrum antibiotic does not impair maternal care in the European earwig | Sophie Van Meyel, Séverine Devers, Simon Dupont, Franck Dedeine and Joël Meunier | <p>The microbes residing within the gut of an animal host often increase their own fitness by modifying their host’s physiological, reproductive, and behavioural functions. Whereas recent studies suggest that they may also shape host sociality and... | | Behavior & Social Evolution, Evolutionary Ecology, Experimental Evolution, Life History, Species interactions | Trine Bilde | 2020-10-09 14:07:47 | ||
15 Dec 2016

POSTPRINT
Basidiomycete yeasts in the cortex of ascomycete macrolichensNew partner at the core of macrolichen diversityRecommended by Enric Frago and Benoit FaconIt has long been known that most multicellular eukaryotes rely on microbial partners for a variety of functions including nutrition, immune reactions and defence against enemies. Lichens are probably the most popular example of a symbiosis involving a photosynthetic microorganism (an algae, a cyanobacteria or both) living embedded within the filaments of a fungus (usually an ascomycete). The latter is the backbone structure of the lichen, whereas the former provides photosynthetic products. Lichens are unique among symbioses because the structures the fungus and the photosynthetic microorganism form together do not resemble any of the two species living in isolation. Classic textbook examples like lichens are not often challenged and this is what Toby Spribille and his co-authors did with their paper published in July 2016 in Science [1]. This story started with the study of two species of macrolichens from the class of Lecanoromycetes that are commonly found in the mountains of Montana (US): Bryoria fremontii and B. tortuosa. For more than 90 years, these species have been known to differ in their chemical composition and colour, but studies performed so far failed in finding differences at the molecular level in both the mycobiont and the photobiont. These two species were therefore considered as nomenclatural synonyms, and the origin of their differences remained elusive. To solve this mystery, the authors of this work performed a transcriptome-wide analysis that, relative to previous studies, expanded the taxonomic range to all Fungi. This analysis revealed higher abundances of a previously unknown basidiomycete yeast from the genus Cyphobasidium in one of the lichen species, a pattern that was further confirmed by combining microscopy imaging and the fluorescent in situ hybridisation technique (FISH). Finding out that a previously unknown micro-organism changes the colour and the chemical composition of an organism is surprising but not new. For instance, bacterial symbionts are able to trigger colour changes in some insect species [2], and endophyte fungi are responsible for the production of defensive compounds in the leaves of several grasses [3]. The study by Spribille and his co-authors is fascinating because it demonstrates that Cyphobasidium yeasts have played a key role in the evolution and diversification of Lecanoromycetes, one of the most diverse classes of macrolichens. Indeed these basidiomycete yeasts were not only found in Bryoria but in 52 other lichen genera from all six continents, and these included 42 out of 56 genera in the family Parmeliaceae. Most of these sequences formed a highly supported monophyletic group, and a molecular clock revealed that the origin of many macrolichen groups occurred around the same time Cyphobasidium yeasts split from Cystobasidium, their nearest relatives. This newly discovered passenger is therefore an ancient inhabitant of lichens and has driven the evolution of this emblematic group of organisms. This study raises an important question on the stability of complex symbiotic partnerships. In intimate obligatory symbioses the evolutionary interests of both partners are often identical and what is good for one is also good for the other. This is the case of several insects that feed on poor diets like phloem and xylem sap, and which carry vertically-transmitted symbionts that provide essential nutrients. Molecular phylogenetic studies have repeatedly shown that in several insect groups transition to phloem or xylem feeding occurred at the same time these nutritional symbionts were acquired [4]. In lichens, an outstanding question is to know what was the key feature Cyphobasidium yeasts brought to the symbiosis. As suggested by the authors, these yeasts are likely to be involved in the production of secondary defensive metabolites and architectural structures, but, are these services enough to explain the diversity found in macrolichens? This paper is an appealing example of a multipartite symbiosis where the different partners share an ancient evolutionary history. References [1] Spribille T, Tuovinen V, Resl P, et al. 2016. Basidiomycete yeasts in the cortex of ascomycete macrolichens. Science 353:488–92. doi: 10.1126/science.aaf8287 [2] Tsuchida T, Koga R, Horikawa M, et al. 2010. Symbiotic Bacterium Modifies Aphid Body Color. Science 330:1102–1104. doi: 10.1126/science.1195463 [3] Clay K. 1988. Fungal Endophytes of Grasses: A Defensive Mutualism between Plants and Fungi. Ecology 69:10–16. doi: 10.2307/1943155 [4] Moran NA. 2007. Symbiosis as an adaptive process and source of phenotypic complexity. Proceeding of the National Academy of Science USA 104:8627–8633. doi: 10.1073/pnas.0611659104 | Basidiomycete yeasts in the cortex of ascomycete macrolichens | Spribille T, Tuovinen V, Resl P, et al. | For over 140 years, lichens have been regarded as a symbiosis between a single fungus, usually an ascomycete, and a photosynthesizing partner. Other fungi have long been known to occur as occasional parasites or endophytes, but the one lichen–one ... | | Adaptation, Evolutionary Ecology, Genome Evolution, Genotype-Phenotype, Life History, Macroevolution, Molecular Evolution, Phylogenetics / Phylogenomics, Speciation, Species interactions | Enric Frago | 2016-12-15 05:46:14 |




