
Latest recommendations

| Id | Title | Authors▲ | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
02 Sep 2022

Introgression between highly divergent sea squirt genomes: an adaptive breakthrough?A match made in the Anthropocene: human-mediated adaptive introgression across a speciation continuumRecommended by Fernando Racimo based on reviews by Michael Westbury, Andrew Foote and Erin CalfeeThe long-distance transport and introduction of new species by humans is increasingly leading divergent lineages to interact, and sometimes interbreed, even after thousands or millions of years of separation. It is thus of prime importance to understand the consequences of these contemporary admixture events on the evolutionary fitness of interacting organisms, and their ecological implications. Ciona robusta and Ciona intestinalis are two species of sea squirts that diverged between 1.5 and 2 million years ago and recently came into contact again. This occurred through human-mediated introduction of C. robusta (native to the Northwest Pacific) into the range of C. intestinalis (the English channeled Northeast Atlantic). In this study, Fraïsse et al. (2022) follow up on earlier work by Le Moan et al. (2021), who had identified a long genomic hotspot of introgression of C. robusta ancestry segments in chromosome 5 of C. intestinalis. The hotspot bears suggestive evidence of positive selection and the authors aimed to investigate this further using fully phased whole-genome sequences. The authors narrow down on the exact boundaries of the introgressed region, and make a compelling case that it has been the likely target of positive selection after introgression, using various complementary approaches based on patterns of population differentiation, haplotype structure and local levels of diversity in the region. Using extensive demographic modeling, they also show that the introgression event was likely recent (approximately 75 years ago), and distinct from other tracts in the C. intestinalis genome that are likely a product of more ancient episodes of interbreeding in the past 30,000 years. Narrowing down on the potential drivers of selection, the authors show that candidate SNPs in the region overlap with the cytochrome family 2 subfamily U gene - involved in the detoxification of exogenous compounds - potentially reflecting adaptation to chemicals encountered in the sea squirt's environment. There also appears to be copy number variation at the candidate SNPs, which provides clues into the adaptation mechanism in the region. All reviewers agreed that the work carried out by the authors is elegant and the results are robustly supported and well presented. In a round of reviews, various clarifications of the manuscript were suggested by the reviewers, including on the quality of the newly generated sequencing data, and some suggestions for qualifications on the conclusions reached by the authors as well as changes in the figures to increase their clarity. The authors addressed the different concerns of the reviewers, and the new version is much improved. This study into human-mediated introgression and its consequences for adaptation is, in my view, both well thought-out and executed. I therefore provide an enthusiastic recommendation of this manuscript. References Fraïsse C, Le Moan A, Roux C, Dubois G, Daguin-Thiébaut C, Gagnaire P-A, Viard F and Bierne N (2022) Introgression between highly divergent sea squirt genomes: an adaptive breakthrough? bioRxiv, 2022.03.22.485319, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.03.22.485319 Le Moan A, Roby C, Fraïsse C, Daguin-Thiébaut C, Bierne N, Viard F (2021) An introgression breakthrough left by an anthropogenic contact between two ascidians. Molecular Ecology, 30, 6718–6732. https://doi.org/10.1111/mec.16189 | Introgression between highly divergent sea squirt genomes: an adaptive breakthrough? | Christelle Fraïsse, Alan Le Moan, Camille Roux, Guillaume Dubois, Claire Daguin-Thiébaut, Pierre-Alexandre Gagnaire, Frédérique Viard, Nicolas Bierne | <p style="text-align: justify;">Human-mediated introductions are reshuffling species distribution on a global scale. Consequently, an increasing number of allopatric taxa are now brought into contact, promoting introgressive hybridization between ... | | Adaptation, Hybridization / Introgression, Population Genetics / Genomics | Fernando Racimo | 2022-04-14 15:30:42 | ||
16 Nov 2018

Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen IslandsIntrogression from related species reveals fine-scale structure in an isolated population of mussels and causes patterns of genetic-environment associationsRecommended by Marianne Elias based on reviews by Thomas Broquet and Tatiana GiraudAssessing population connectivity is central to understanding population dynamics, and is therefore of great importance in evolutionary biology and conservation biology. In the marine realm, the apparent absence of physical barriers, large population sizes and high dispersal capacities of most organisms often result in no detectable structure, thereby hindering inferences of population connectivity. In a review paper, Gagnaire et al. [1] propose several ideas to improve detection of population connectivity. Notably, using simulations they show that under certain circumstances introgression from one species into another may reveal cryptic population structure within that second species. References [1] Gagnaire, P.-A., Broquet, T., Aurelle, D., Viard, F., Souissi, A., Bonhomme, F., Arnaud-Haond, S., & Bierne, N. (2015). Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evolutionary Applications, 8, 769–786. doi: 10.1111/eva.12288 | Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands | Christelle Fraïsse, Anne Haguenauer, Karin Gerard, Alexandra Anh-Thu Weber, Nicolas Bierne, Anne Chenuil | <p>Reticulated evolution -i.e. secondary introgression / admixture between sister taxa- is increasingly recognized as playing a key role in structuring infra-specific genetic variation and revealing cryptic genetic connectivity patterns. When admi... | | Hybridization / Introgression, Phylogeography & Biogeography, Population Genetics / Genomics | Marianne Elias | 2017-12-28 14:16:16 | ||
30 Jun 2023
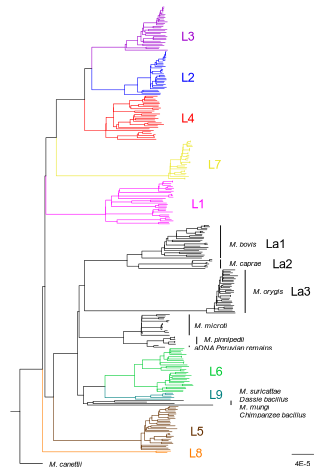
How do monomorphic bacteria evolve? The Mycobacterium tuberculosis complex and the awkward population genetics of extreme clonalityHow the tubercle bacillus got its genome: modernising, modelling, and making sense of the stories we tellRecommended by B. Jesse Shapiro based on reviews by 2 anonymous reviewersIn this instructive review, Stritt and Gagneux offer a balanced perspective on the evolutionary forces shaping Mycobacterium tuberculosis and make the case that our instinct for storytelling be balanced with quantitative models. M. tuberculosis is perhaps the best-known clonal bacterial pathogen – evolving largely in the absence of horizontal gene transfer. Its genome is full of puzzling patterns, including much higher GC content than most intracellular pathogens (which suggests efficient selection to resist AT-skewed mutational bias) but a very high ratio of nonsynonymous to synonymous substitution rates (dN/dS ~ 0.5, typically interpreted as weak selection against deleterious amino acid changes). The authors offer alternative explanations for these patterns, framing the question: is M. tuberculosis evolution shaped mainly by drift or by efficient selection? They propose that this question can only be answered by accounting for the pathogen’s extreme clonality. A clonal lifestyle can have its advantages, for example when adaptations must arise in a particular order (Kondrashov and Kondrashov 2001). An important disadvantage highlighted by the authors are linkage effects: without recombination to shuffle them up, beneficial mutations are linked to deleterious mutations in the same genome (hitchhiking) and purging deleterious mutations also purges neutral diversity across the genome (background selection). The authors propose the latter – efficient purifying selection and strong linkage – as an explanation for the low genetic diversity observed in M. tuberculosis. This is of course not exclusive of other related explanations, such as clonal interference (Gerrish and Lenski 1998). They also champion the use of forward evolutionary simulations (Haller and Messer 2019) to model the interplay between selection, recombination, and demography as a powerful alternative to traditional backward coalescent models. At times, Stritt and Gagneux are pessimistic about our existing methods – arguing that dN/dS and homoplasies “tell us little about the frequency and strength of selection.” Even though I favour a more optimistic view, I fully agree that our traditional population genetic metrics are sensitive to a slew of different deviations from a standard neutral evolution model and must be interpreted with caution. As I and others have argued, the extent of recombination (measured as the amount of linkage in a genome) is a key factor in determining how best to test for natural selection (Shapiro et al. 2009) and to conduct genotype-phenotype association studies (Chen and Shapiro 2021) in microbes. While this article is focused on the well-studied M. tuberculosis complex, there are many parallels with other clonal bacteria, including pathogens and symbionts. Whatever your favourite bug, we must all be careful to make sure the stories we tell about them are not “just so tales” but are supported, to the extent possible, by data and quantitative models. References Chen, Peter E., and B. Jesse Shapiro. 2021. "Classic Genome-Wide Association Methods Are Unlikely to Identify Causal Variants in Strongly Clonal Microbial Populations." bioRxiv. Stritt, C., Gagneux, S. (2023). How do monomorphic bacteria evolve? The Mycobacterium tuberculosis complex and the awkward population genetics of extreme clonality. EcoEvoRxiv, ver.3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.32942/X2GW2P | How do monomorphic bacteria evolve? The *Mycobacterium tuberculosis* complex and the awkward population genetics of extreme clonality | Christoph Stritt, Sebastien Gagneux | <p style="text-align: justify;">Exchange of genetic material through sexual reproduction or horizontal gene transfer is ubiquitous in nature. Among the few outliers that rarely recombine and mainly evolve by <em>de novo</em> mutation are a group o... | | Evolutionary Dynamics, Genome Evolution, Molecular Evolution, Population Genetics / Genomics, Reproduction and Sex | B. Jesse Shapiro | Gonçalo Themudo | 2022-12-16 13:41:53 | |
17 Jun 2022

Spontaneous parthenogenesis in the parasitoid wasp Cotesia typhae: low frequency anomaly or evolving process?The potential evolutionary importance of low-frequency flexibility in reproductive modesRecommended by Christoph Haag based on reviews by Michael Lattorff and Jens BastOccasional events of asexual reproduction in otherwise sexual taxa have been documented since a long time. Accounts range from observations of offspring development from unfertilized eggs in Drosophila to rare offspring production by isolated females in lizards and birds (e.g., Stalker 1954, Watts et al 2006, Ryder et al. 2021). Many more such cases likely await documentation, as rare events are inherently difficult to observe. These rare events of asexual reproduction are often associated with low offspring fitness (“tychoparthenogenesis”), and have mostly been discarded in the evolutionary literature as reproductive accidents without evolutionary significance. Recently, however, there has been an increased interest in the details of evolutionary transitions from sexual to asexual reproduction (e.g., Archetti 2010, Neiman et al.2014, Lenormand et al. 2016), because these details may be key to understanding why successful transitions are rare, why they occur more frequently in some groups than in others, and why certain genetic mechanisms of ploidy maintenance or ploidy restoration are more often observed than others. In this context, the hypothesis has been formulated that regular or even obligate asexual reproduction may evolve from these rare events of asexual reproduction (e.g., Schwander et al. 2010). A new study by Capdevielle Dulac et al. (2022) now investigates this question in a parasitoid wasp, highlighting also the fact that what is considered rare or occasional may differ from one system to the next. The results show “rare” parthenogenetic production of diploid daughters occurring at variable frequencies (from zero to 2 %) in different laboratory strains, as well as in a natural population. They also demonstrate parthenogenetic production of female offspring in both virgin females and mated ones, as well as no reduced fecundity of parthenogenetically produced offspring. These findings suggest that parthenogenetic production of daughters, while still being rare, may be a more regular and less deleterious reproductive feature in this species than in other cases of occasional asexuality. Indeed, haplodiploid organisms, such as this parasitoid wasp have been hypothesized to facilitate evolutionary transitions to asexuality (Neimann et al. 2014, Van Der Kooi et al. 2017). First, in haploidiploid organisms, females are diploid and develop from normal, fertilized eggs, but males are haploid as they develop parthenogenetically from unfertilized eggs. This means that, in these species, fertilization is not necessarily needed to trigger development, thus removing one of the constraints for transitions to obligate asexuality (Engelstädter 2008, Vorburger 2014). Second, spermatogenesis in males occurs by a modified meiosis that skips the first meiotic division (e.g., Ferree et al. 2019). Haploidiploid organisms may thus have a potential route for an evolutionary transition to obligate parthenogenesis that is not available to organisms: The pathways for the modified meiosis may be re-used for oogenesis, which might result in unreduced, diploid eggs. Third, the particular species studied here regularly undergoes inbreeding by brother-sister mating within their hosts. Homozygosity, including at the sex determination locus (Engelstädter 2008), is therefore expected to have less negative effects in this species compared to many other, non-inbreeding haplodipoids (see also Little et al. 2017). This particular species may therefore be less affected by loss of heterozygosity, which occurs in a fashion similar to self-fertilization under many forms of non-clonal parthenogenesis. Indeed, the study also addresses the mechanisms underlying parthenogenesis in the species. Surprisingly, the authors find that parthenogenetically produced females are likely produced by two distinct genetic mechanisms. The first results in clonality (maintenance of the maternal genotype), whereas the second one results in a loss of heterozygosity towards the telomeres, likely due to crossovers occurring between the centromeres and the telomeres. Moreover, bacterial infections appear to affect the propensity of parthenogenesis but are unlikely the primary cause. Together, the finding suggests that parthenogenesis is a variable trait in the species, both in terms of frequency and mechanisms. It is not entirely clear to what degree this variation is heritable, but if it is, then these results constitute evidence for low-frequency existence of variable and heritable parthenogenesis phenotypes, that is, the raw material from which evolutionary transitions to more regular forms of parthenogenesis may occur.
References Archetti M (2010) Complementation, Genetic Conflict, and the Evolution of Sex and Recombination. Journal of Heredity, 101, S21–S33. https://doi.org/10.1093/jhered/esq009 Capdevielle Dulac C, Benoist R, Paquet S, Calatayud P-A, Obonyo J, Kaiser L, Mougel F (2022) Spontaneous parthenogenesis in the parasitoid wasp Cotesia typhae: low frequency anomaly or evolving process? bioRxiv, 2021.12.13.472356, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.12.13.472356 Engelstädter J (2008) Constraints on the evolution of asexual reproduction. BioEssays, 30, 1138–1150. https://doi.org/10.1002/bies.20833 Ferree PM, Aldrich JC, Jing XA, Norwood CT, Van Schaick MR, Cheema MS, Ausió J, Gowen BE (2019) Spermatogenesis in haploid males of the jewel wasp Nasonia vitripennis. Scientific Reports, 9, 12194. https://doi.org/10.1038/s41598-019-48332-9 van der Kooi CJ, Matthey-Doret C, Schwander T (2017) Evolution and comparative ecology of parthenogenesis in haplodiploid arthropods. Evolution Letters, 1, 304–316. https://doi.org/10.1002/evl3.30 Lenormand T, Engelstädter J, Johnston SE, Wijnker E, Haag CR (2016) Evolutionary mysteries in meiosis. Philosophical Transactions of the Royal Society B: Biological Sciences, 371, 20160001. https://doi.org/10.1098/rstb.2016.0001 Little CJ, Chapuis M-P, Blondin L, Chapuis E, Jourdan-Pineau H (2017) Exploring the relationship between tychoparthenogenesis and inbreeding depression in the Desert Locust, Schistocerca gregaria. Ecology and Evolution, 7, 6003–6011. https://doi.org/10.1002/ece3.3103 Neiman M, Sharbel TF, Schwander T (2014) Genetic causes of transitions from sexual reproduction to asexuality in plants and animals. Journal of Evolutionary Biology, 27, 1346–1359. https://doi.org/10.1111/jeb.12357 Ryder OA, Thomas S, Judson JM, Romanov MN, Dandekar S, Papp JC, Sidak-Loftis LC, Walker K, Stalis IH, Mace M, Steiner CC, Chemnick LG (2021) Facultative Parthenogenesis in California Condors. Journal of Heredity, 112, 569–574. https://doi.org/10.1093/jhered/esab052 Schwander T, Vuilleumier S, Dubman J, Crespi BJ (2010) Positive feedback in the transition from sexual reproduction to parthenogenesis. Proceedings of the Royal Society B: Biological Sciences, 277, 1435–1442. https://doi.org/10.1098/rspb.2009.2113 Stalker HD (1954) Parthenogenesis in Drosophila. Genetics, 39, 4–34. https://doi.org/10.1093/genetics/39.1.4 Vorburger C (2014) Thelytoky and Sex Determination in the Hymenoptera: Mutual Constraints. Sexual Development, 8, 50–58. https://doi.org/10.1159/000356508 Watts PC, Buley KR, Sanderson S, Boardman W, Ciofi C, Gibson R (2006) Parthenogenesis in Komodo dragons. Nature, 444, 1021–1022. https://doi.org/10.1038/4441021a | Spontaneous parthenogenesis in the parasitoid wasp Cotesia typhae: low frequency anomaly or evolving process? | Claire Capdevielle Dulac, Romain Benoist, Sarah Paquet, Paul-André Calatayud, Julius Obonyo, Laure Kaiser, Florence Mougel | <p style="text-align: justify;">Hymenopterans are haplodiploids and unlike most other Arthropods they do not possess sexual chromosomes. Sex determination typically happens via the ploidy of individuals: haploids become males and diploids become f... | | Evolutionary Ecology, Life History, Reproduction and Sex | Christoph Haag | 2021-12-16 15:25:16 | ||
13 Dec 2016
POSTPRINT
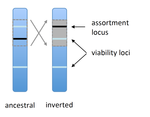
Prezygotic isolation, mating preferences, and the evolution of chromosomal inversionsThe spread of chromosomal inversions as a mechanism for reinforcementRecommended by Denis Roze and Thomas Broquet
Several examples of chromosomal inversions carrying genes affecting mate choice have been reported from various organisms. Furthermore, inversions are also frequently involved in genetic isolation between populations or species. Past work has shown that inversions can spread when they capture not only some loci involved in mate choice but also loci involved in incompatibilities between hybridizing populations [1]. In this new paper [2], the authors derive analytical approximations for the selection coefficient associated with an inversion suppressing recombination between a locus involved in mate choice and one (or several) locus involved in Dobzhansky-Muller incompatibilities. Two mechanisms for mate choice are considered: assortative mating based on the allele present at a single locus, or a trait-preference model where one locus codes for the trait and another for the preference. The results show that such an inversion is generally favoured, the selective advantage associated with the inversion being strongest when hybridization is sufficiently frequent. Assuming pairwise epistatic interactions between loci involved in incompatibilities, selection for the inversion increases approximately linearly with the number of such loci captured by the inversion. This paper is a good read for several reasons. First, it presents the problem clearly (e.g. the introduction provides a clear and concise presentation of the issue and past work) and its crystal-clear writing facilitates the reader's understanding of theoretical approaches and results. Second, the analysis is competently done and adds to previous work by showing that very general conditions are expected to be favourable to the spread of the type of inversion considered here. And third, it provides food for thought about the role of inversions in the origin or the reinforcement of divergence between nascent species. One result of this work is that an inversion linked to pre-zygotic isolation "is favoured so long as there is viability selection against recombinant genotypes", suggesting that genetic incompatibilities must have evolved first and that inversions capturing mating preference loci may then enhance pre-existing reproductive isolation. However, the results also show that inversions are more likely to be favoured in hybridizing populations among which gene flow is still high, rather than in more strongly isolated populations. This matches the observation that inversions are more frequently observed between sympatric species than between allopatric ones. References [1] Trickett AJ, Butlin RK. 1994. Recombination Suppressors and the Evolution of New Species. Heredity 73:339-345. doi: 10.1038/hdy.1994.180 [2] Dagilis AJ, Kirkpatrick M. 2016. Prezygotic isolation, mating preferences, and the evolution of chromosomal inversions. Evolution 70: 1465–1472. doi: 10.1111/evo.12954 | Prezygotic isolation, mating preferences, and the evolution of chromosomal inversions | Dagilis AJ, Kirkpatrick M | Chromosomal inversions are frequently implicated in isolating species. Models have shown how inversions can evolve in the context of postmating isolation. Inversions are also frequently associated with mating preferences, a topic that has not been... | | Adaptation, Evolutionary Theory, Genome Evolution, Hybridization / Introgression, Population Genetics / Genomics, Speciation | Denis Roze | 2016-12-13 22:11:54 | ||
02 Nov 2020

Experimental evolution of virulence and associated traits in a Drosophila melanogaster – Wolbachia symbiosisTemperature effects on virulence evolution of wMelPop Wolbachia in Drosophila melanogasterRecommended by Ellen Decaestecker based on reviews by Shira Houwenhuyse and 3 anonymous reviewersMonnin et al. [1] here studied how Drosophila populations are affected when exposed to a high virulent endosymbiotic wMelPop Wolbachia strain and why virulent vertically transmitting endosymbionts persist in nature. This virulent wMelPop strain has been described to be a blocker of dengue and other arboviral infections in arthropod vector species, such as Aedes aegypti. Whereas it can thus function as a mutualistic symbiont, it here acts as an antagonist along the mutualism-antagonism continuum symbionts operate. The wMelPop strain is not a natural occurring strain in Drosophila melanogaster and thus the start of this experiment can be seen as a novel host-pathogen association. Through experimental evolution of 17 generations, the authors studied how high temperature affects wMelPop Wolbachia virulence and Drosophila melanogaster survival. The authors used Drosophila strains that were selected for late reproduction, given that this should favor evolution to a lower virulence. Assumptions for this hypothesis are not given in the manuscript here, but it can indeed be assumed that energy that is assimilated to symbiont tolerance instead of reproduction may lead to reduced virulence evolution. This has equally been suggested by Reyserhove et al. [2] in a dynamics energy budget model tailored to Daphnia magna virulence evolution upon a viral infection causing White fat Cell disease, reconstructing changing environments through time. References [1] Monnin, D., Kremer, N., Michaud, C., Villa, M., Henri, H., Desouhant, E. and Vavre, F. (2020) Experimental evolution of virulence and associated traits in a Drosophila melanogaster – Wolbachia symbiosis. bioRxiv, 2020.04.26.062265, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: https://doi.org/10.1101/2020.04.26.062265 | Experimental evolution of virulence and associated traits in a Drosophila melanogaster – Wolbachia symbiosis | David Monnin, Natacha Kremer, Caroline Michaud, Manon Villa, Hélène Henri, Emmanuel Desouhant, Fabrice Vavre | <p>Evolutionary theory predicts that vertically transmitted symbionts are selected for low virulence, as their fitness is directly correlated to that of their host. In contrast with this prediction, the Wolbachia strain wMelPop drastically reduces... | | Evolutionary Ecology, Experimental Evolution, Species interactions | Ellen Decaestecker | 2020-04-29 19:16:56 | ||
08 Jan 2024
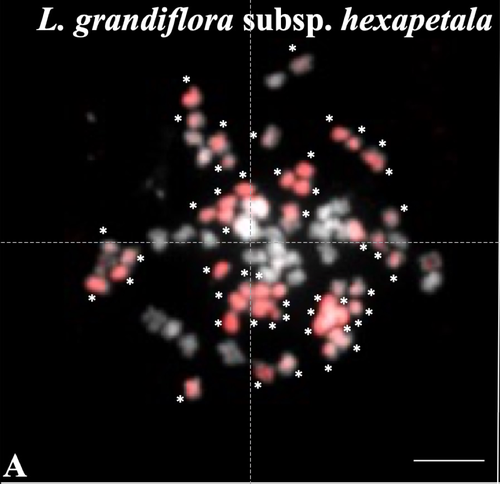
Genomic relationships among diploid and polyploid species of the genus Ludwigia L. section Jussiaea using a combination of molecular cytogenetic, morphological, and crossing investigationsDeciphering the genomic composition of tetraploid, hexaploid and decaploid Ludwigia L. species (section Jussiaea)Recommended by Malika AINOUCHE based on reviews by Alex BAUMEL and Karol MARHOLDPolyploidy, which results in the presence of more than two sets of homologous chromosomes represents a major feature of plant genomes that have undergone successive rounds of duplication followed by more or less rapid diploidization during their evolutionary history. Polyploid complexes containing diploid and derived polyploid taxa are excellent model systems for understanding the short-term consequences of whole genome duplication, and have been particularly well-explored in evolutionary ecology (Ramsey and Ramsey 2014, Rice et al. 2019). Many polyploids (especially when resulting from interspecific hybridization, i.e. allopolyploids) are successful invaders (te Beest et al. 2012) as a result of rapid genome dynamics, functional novelty, and trait evolution. The origin (parental legacy) and modes of formation of polyploids have a critical impact on the subsequent polyploid evolution. Thus, elucidation of the genomic composition of polyploids is fundamental to understanding trait evolution, and such knowledge is still lacking for many invasive species. Genus Ludwigia is characterized by a complex taxonomy, with an underexplored evolutionary history. Species from section Jussieae form a polyploid complex with diploids, tetraploids, hexaploids, and decaploids that are notorious invaders in freshwater and riparian ecosystems (Thouvenot et al.2013). Molecular phylogeny of the genus based on nuclear and chloroplast sequences (Liu et al. 2027) suggested some relationships between diploid and polyploid species, without fully resolving the question of the parentage of the polyploids. In their study, Barloy et al. (2023) have used a combination of molecular cytogenetics (Genomic In situ Hybridization), morphology and experimental crosses to elucidate the genomic compositions of the polyploid species, and show that the examined polyploids are of hybrid origin (allopolyploids). The tetraploid L. stolonifera derives from the diploids L. peploides subsp. montevidensis (AA genome) and L. helminthorhiza (BB genome). The tetraploid L. ascendens also share the BB genome combined with an undetermined different genome. The hexaploid L. grandiflora subsp. grandiflora has inherited the diploid AA genome combined with additional unidentified genomes. The decaploid L. grandiflora subsp. hexapetala has inherited the tetraploid L. stolonifera and the hexaploid L. grandiflora subsp. hexapetala genomes. As the authors point out, further work is needed, including additional related diploid (e.g. other subspecies of L. peploides) or tetraploid (L. hookeri and L. peduncularis) taxa that remain to be investigated, to address the nature of the undetermined parental genomes mentioned above. The presented work (Barloy et al. 2023) provides significant knowledge of this poorly investigated group with regard to genomic information and polyploid origin, and opens perspectives for future studies. The authors also detect additional diagnostic morphological traits of interest for in-situ discrimination of the taxa when monitoring invasive populations. References Barloy D., Portillo-Lemus L., Krueger-Hadfield S.A., Huteau V., Coriton O. (2024). Genomic relationships among diploid and polyploid species of the genus Ludwigia L. section Jussiaea using a combination of molecular cytogenetic, morphological, and crossing investigations. BioRxiv, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology https://doi.org/10.1101/2023.01.02.522458 te Beest M., Le Roux J.J., Richardson D.M., Brysting A.K., Suda J., Kubešová M., Pyšek P. (2012). The more the better? The role of polyploidy in facilitating plant invasions. Annals of Botany, Volume 109, Issue 1 Pages 19–45, https://doi.org/10.1093/aob/mcr277 Ramsey J. and Ramsey T. S. (2014). Ecological studies of polyploidy in the 100 years following its discovery Phil. Trans. R. Soc. B369 1–20 https://doi.org/10.1098/rstb.2013.0352 Rice, A., Šmarda, P., Novosolov, M. et al. (2019). The global biogeography of polyploid plants. Nat Ecol Evol 3, 265–273. https://doi.org/10.1038/s41559-018-0787-9 Thouvenot L, Haury J, Thiebaut G. (2013). A success story: Water primroses, aquatic plant pests. Aquat. Conserv. Mar. Freshw. Ecosyst. 23:790–803 https://doi.org/10.1002/aqc.2387 | Genomic relationships among diploid and polyploid species of the genus *Ludwigia* L. section *Jussiaea* using a combination of molecular cytogenetic, morphological, and crossing investigations | D. Barloy, L. Portillo - Lemus, S. A. Krueger-Hadfield, V. Huteau, O. Coriton | <p>ABSTRACTThe genus Ludwigia L. sectionJussiaeais composed of a polyploid species complex with 2x, 4x, 6x and 10x ploidy levels, suggesting possible hybrid origins. The aim of the present study is to understand the genomic relationships among dip... | | Hybridization / Introgression, Phylogenetics / Phylogenomics | Malika AINOUCHE | 2023-01-11 13:47:18 | ||
13 Dec 2018

A behavior-manipulating virus relative as a source of adaptive genes for parasitoid waspsGenetic intimacy of filamentous viruses and endoparasitoid waspsRecommended by Ignacio Bravo based on reviews by Alejandro Manzano Marín and 1 anonymous reviewerViruses establish intimate relationships with the cells they infect. The virocell is a novel entity, different from the original host cell and beyond the mere combination of viral and cellular genetic material. In these close encounters, viral and cellular genomes often hybridise, combine, recombine, merge and excise. Such chemical promiscuity leaves genomics scars that can be passed on to descent, in the form of deletions or duplications and, importantly, insertions and back and forth exchange of genetic material between viruses and their hosts. References [1] Di Giovanni, D., Lepetit, D., Boulesteix, M., Ravallec, M., & Varaldi, J. (2018). A behavior-manipulating virus relative as a source of adaptive genes for parasitoid wasps. bioRxiv, 342758, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/342758 | A behavior-manipulating virus relative as a source of adaptive genes for parasitoid wasps | D. Di Giovanni, D. Lepetit, M. Boulesteix, M. Ravallec, J. Varaldi | <p>To circumvent host immune response, numerous hymenopteran endo-parasitoid species produce virus-like structures in their reproductive apparatus that are injected into the host together with the eggs. These viral-like structures are absolutely n... | | Adaptation, Behavior & Social Evolution, Genetic conflicts, Genome Evolution | Ignacio Bravo | 2018-07-18 15:59:14 | ||
20 Nov 2017
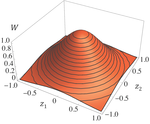
Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selectionUnderstanding genetic variance, load, and inbreeding depression with selfingRecommended by Aneil F. Agrawal based on reviews by Frédéric Guillaume and 1 anonymous reviewerA classic problem in evolutionary biology is to understand the genetic variance in fitness. The simplest hypothesis is that variation exists, even in well-adapted populations, as a result of the balance between mutational input and selective elimination. This variation causes a reduction in mean fitness, known as the mutation load. Though mutation load is difficult to quantify empirically, indirect evidence of segregating genetic variation in fitness is often readily obtained by comparing the fitness of inbred and outbred offspring, i.e., by measuring inbreeding depression. Mutation-selection balance models have been studied as a means of understanding the genetic variance in fitness, mutation load, and inbreeding depression. Since their inception, such models have increased in sophistication, allowing us to ask these questions under more realistic and varied scenarios. The new theoretical work by Abu Awad and Roze [1] is a substantial step forward in understanding how arbitrary levels of self-fertilization affect variation, load and inbreeding depression under mutation-selection balance. References [1] Abu Awad D and Roze D. 2017. Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selection. bioRxiv, 180000, ver. 4 of 17th November 2017. doi: 10.1101/180000 [2] Lande R. 1977. The influence of the mating system on the maintenance of genetic variability in polygenic characters. Genetics 86: 485–498. [3] Charlesworth D and Charlesworth B. 1987. Inbreeding depression and its evolutionary consequences. Annual Review of Ecology and Systematics. 18: 237–268. doi: 10.1111/10.1146/annurev.es.18.110187.001321 [4] Lande R and Porcher E. 2015. Maintenance of quantitative genetic variance under partial self-fertilization, with implications for the evolution of selfing. Genetics 200: 891–906. doi: 10.1534/genetics.115.176693 [5] Roze D. 2015. Effects of interference between selected loci on the mutation load, inbreeding depression, and heterosis. Genetics 201: 745–757. doi: 10.1534/genetics.115.178533 [6] Martin G and Lenormand T. 2006. A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species. Evolution 60: 893–907. doi: 10.1111/j.0014-3820.2006.tb01169.x [7] Martin G, Elena SF and Lenormand T. 2007. Distributions of epistasis in microbes fit predictions from a fitness landscape model. Nature Genetics 39: 555–560. doi: 10.1038/ng1998 | Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selection | Diala Abu Awad and Denis Roze | The mating system of a species is expected to have important effects on its genetic diversity. In this paper, we explore the effects of partial selfing on the equilibrium genetic variance Vg, mutation load L and inbreeding depression δ under stabi... | | Evolutionary Theory, Population Genetics / Genomics, Quantitative Genetics, Reproduction and Sex | Aneil F. Agrawal | 2017-08-26 09:29:20 | ||
17 Feb 2020
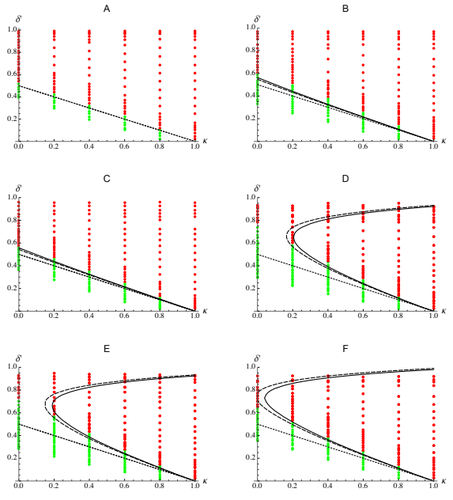
Epistasis, inbreeding depression and the evolution of self-fertilizationEpistasis and the evolution of selfingRecommended by Sylvain Gandon based on reviews by Nick Barton and 1 anonymous reviewerThe evolution of selfing results from a balance between multiple evolutionary forces. Selfing provides an "automatic advantage" due to the higher efficiency of selfers to transmit their genes via selfed and outcrossed offspring. Selfed offspring, however, may suffer from inbreeding depression. In principle the ultimate evolutionary outcome is easy to predict from the relative magnitude of these two evolutionary forces [1,2]. Yet, several studies explicitly taking into account the genetic architecture of inbreeding depression noted that these predictions are often too restrictive because selfing can evolve in a broader range of conditions [3,4]. References [1] Holsinger, K. E., Feldman, M. W., and Christiansen, F. B. (1984). The evolution of self-fertilization in plants: a population genetic model. The American Naturalist, 124(3), 446-453. doi: 10.1086/284287 | Epistasis, inbreeding depression and the evolution of self-fertilization | Diala Abu Awad and Denis Roze | <p>Inbreeding depression resulting from partially recessive deleterious alleles is thought to be the main genetic factor preventing self-fertilizing mutants from spreading in outcrossing hermaphroditic populations. However, deleterious alleles may... | | Evolutionary Theory, Quantitative Genetics, Reproduction and Sex | Sylvain Gandon | 2019-10-18 09:29:41 |




