
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields | Recommender▲ | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
22 Mar 2022
Substantial genetic mixing among sexual and androgenetic lineages within the clam genus CorbiculaStrange reproductive modes and population geneticsRecommended by Chris Jiggins based on reviews by Arnaud Estoup, Simon Henry Martin and 2 anonymous reviewersThere are many organisms that are asexual or have unusual modes of reproduction. One such quasi-sexual reproductive mode is androgenesis, in which the offspring, after fertilization, inherits only the entire paternal nuclear genome. The maternal genome is ditched along the way. One group of organisms which shows this mode of reproduction are clams in the genus Corbicula, some of which are androecious, while others are dioecious and sexual. The study by Vastrade et al. (2022) describes population genetic patterns in these clams, using both nuclear and mitochondrial sequence markers. In contrast to what might be expected for an asexual lineage, there is evidence for significant genetic mixing between populations. In addition, there is high heterozygosity and evidence for polyploidy in some lineages. Overall, the picture is complicated! However, what is clear is that there is far more genetic mixing than expected. One possible mechanism by which this could occur is 'nuclear capture' where there is a mixing of maternal and paternal lineages after fertilization. This can sometimes occur as a result of hybridization between 'species', leading to further mixing of divergent lineages. Thus the group is clearly far from an ancient asexual lineage - recombination and mixing occur with some regularity. The study also analyzed recent invasive populations in Europe and America. These had reduced genetic diversity, but also showed complex patterns of allele sharing suggesting a complex origin of the invasive lineages. In the future, it will be exciting to apply whole genome sequencing approaches to systems such as this. There are challenges in interpreting a handful of sequenced markers especially in a system with polyploidy and considerable complexity, and whole-genome sequencing could clarify some of the outstanding questions, Overall, this paper highlights the complex genetic patterns that can result through unusual reproductive modes, which provides a challenge for the field of population genetics and for the recognition of species boundaries. References Vastrade M, Etoundi E, Bournonville T, Colinet M, Debortoli N, Hedtke SM, Nicolas E, Pigneur L-M, Virgo J, Flot J-F, Marescaux J, Doninck KV (2022) Substantial genetic mixing among sexual and androgenetic lineages within the clam genus Corbicula. bioRxiv, 590836, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/590836 | Substantial genetic mixing among sexual and androgenetic lineages within the clam genus Corbicula | Vastrade M., Etoundi E., Bournonville T., Colinet M., Debortoli N., Hedtke S.M., Nicolas E., Pigneur L.-M., Virgo J., Flot J.-F., Marescaux J. and Van Doninck K. | <p style="text-align: justify;">“Occasional” sexuality occurs when a species combines clonal reproduction and genetic mixing. This strategy is predicted to combine the advantages of both asexuality and sexuality, but its actual consequences on the... | | Evolutionary Ecology, Hybridization / Introgression, Phylogeography & Biogeography | Chris Jiggins | 2019-03-29 15:42:56 | ||
30 Mar 2023
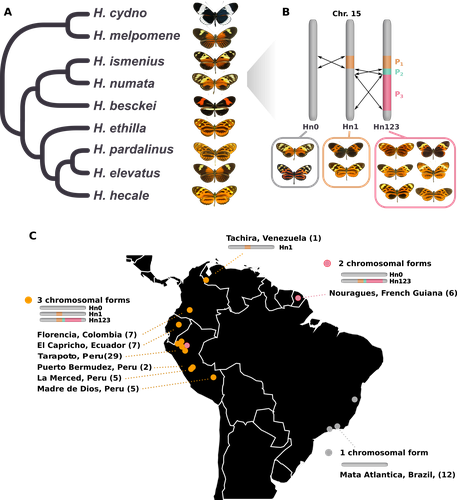
Balancing selection at a wing pattern locus is associated with major shifts in genome-wide patterns of diversity and gene flow in a butterflyIs genetic diversity enhanced by a supergene?Recommended by Chris Jiggins based on reviews by Christelle Fraïsse and 2 anonymous reviewersThe butterfly species Heliconius numata has a remarkable wing pattern polymorphism, with multiple pattern morphs all controlled by a single genetic locus, which harbours multiple inversions. Each morph is a near-perfect mimic of a species in the fairly distantly related genus of butterflies, Melinaea. The article by Rodríguez de Cara et al (2023) argues that the balanced polymorphism at this single wing patterning locus actually has a major effect on genetic diversity across the whole genome. First, polymorphic populations within H. numata are more dioverse than those without polymorphism. Second, H. numata is more genetically diverse than other related species and finally reconstruction of historical demography suggests that there has been a recent increase in effective population size, putatively associated with the acquisition of the supergene polymorphism. The supergene itself generates disassortative mating, such that morphs prefer to mate with others dissimilar to themselves - in this way it is similar to mechanisms for preventing inbreeding such as self-incompatibility loci in plants. This provides a potential mechanism whereby non-random mating patterns could increase effective population size. The authors also explore this mechanism using forward simulations, and show that mating patterns at a single locus can influence linked genetic diversity over a large scale. Overall, this is an intriguing study, which suggests a far more widespread genetic impact of a single locus than might be expected. There are interesting parallels with mechanisms of inbreeding prevention in plants, such as the Pin/Thrum polymorphism in Primula, which also rely on mating patterns determined by a single locus but presumably also influence genetic diversity genome-wide by promoting outbreeding. REFERENCES Rodríguez de Cara MÁ, Jay P, Rougemont Q, Chouteau M, Whibley A, Huber B, Piron-Prunier F, Ramos RR, Freitas AVL, Salazar C, Silva-Brandão KL, Torres TT, Joron M (2023) Balancing selection at a wing pattern locus is associated with major shifts in genome-wide patterns of diversity and gene flow. bioRxiv, 2021.09.29.462348, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.09.29.462348 | Balancing selection at a wing pattern locus is associated with major shifts in genome-wide patterns of diversity and gene flow in a butterfly | María Ángeles Rodríguez de Cara, Paul Jay, Quentin Rougemont, Mathieu Chouteau, Annabel Whibley, Barbara Huber, Florence Piron-Prunier, Renato Rogner Ramos, André V. L. Freitas, Camilo Salazar, Karina Lucas Silva-Brandão, Tatiana Texeira Torres, M... | <p style="text-align: justify;">Selection shapes genetic diversity around target mutations, yet little is known about how selection on specific loci affects the genetic trajectories of populations, including their genomewide patterns of diversity ... | | Evolutionary Ecology, Genome Evolution, Hybridization / Introgression, Population Genetics / Genomics | Chris Jiggins | 2021-10-13 17:54:33 | ||
16 Nov 2022

Divergence of olfactory receptors associated with the evolution of assortative mating and reproductive isolation in miceTinder in mice: A match made with the sense of smellRecommended by Christelle Fraïsse based on reviews by Angeles de Cara, Ludovic Claude Maisonneuve and 1 anonymous reviewer based on reviews by Angeles de Cara, Ludovic Claude Maisonneuve and 1 anonymous reviewer
Differentiation-based genome scans lie at the core of speciation and adaptation genomics research. Dating back to Lewontin & Krakauer (1973), they have become very popular with the advent of genomics to identify genome regions of enhanced differentiation relative to neutral expectations. These regions may represent genetic barriers between divergent lineages and are key for studying reproductive isolation. However, genome scan methods can generate a high rate of false positives, primarily if the neutral population structure is not accounted for (Bierne et al. 2013). Moreover, interpreting genome scans can be challenging in the context of secondary contacts between diverging lineages (Bierne et al. 2011), because the coupling between different components of reproductive isolation (local adaptation, intrinsic incompatibilities, mating preferences, etc.) can occur readily, thus preventing the causes of differentiation from being determined. Smadja and collaborators (2022) applied a sophisticated genome scan for trait association (BAYPASS, Gautier 2015) to underlie the genetic basis of a polygenetic behaviour: assortative mating in hybridizing mice. My interest in this neat study mainly relies on two reasons. First, the authors used an ingenious geographical setting (replicate pairs of “Choosy” versus “Non-Choosy” populations) with multi-way comparisons to narrow down the list of candidate regions resulting from BAYPASS. The latter corrects for population structure, handles cost-effective pool-seq data and allows for gene-based analyses that aggregate SNP signals within a gene. These features reinforce the set of outlier genes detected; however, not all are expected to be associated with mating preference. The second reason why this study is valuable to me is that Smadja et al. (2022) complemented the population genomic approach with functional predictions to validate the genetic signal. In line with previous behavioural and chemical assays on the proximal mechanisms of mating preferences, they identified multiple olfactory and vomeronasal receptor genes as highly significant candidates. Therefore, combining genomic signals with functional analyses is a clever way to provide insights into the causes of reproductive isolation, especially when multiple barriers are involved. This is typically true for reinforcement (Butlin & Smadja 2018), suspected to occur in these mice because, in that case, assortative mating (a prezygotic barrier) evolves in response to the cost of hybridization (for example, due to hybrid inviability). As advocated by the authors, their study paves the way for future work addressing the genetic basis of reinforcement, a trait of major evolutionary importance for which we lack empirical data. They also make a compelling case using complementary approaches that olfactory and vomeronasal receptors have a central role in mammal speciation.
Bierne N, Welch J, Loire E, Bonhomme F, David P (2011) The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol Ecol 20: 2044–2072. https://doi.org/10.1111/j.1365-294X.2011.05080.x Bierne N, Roze D, Welch JJ (2013) Pervasive selection or is it…? why are FST outliers sometimes so frequent? Mol Ecol 22: 2061–2064. https://doi.org/10.1111/mec.12241 Butlin RK, Smadja CM (2018) Coupling, Reinforcement, and Speciation. Am Nat 191:155–172. https://doi.org/10.1086/695136 Gautier M (2015) Genome-Wide Scan for Adaptive Divergence and Association with Population-Specific Covariates. Genetics 201:1555–1579. https://doi.org/10.1534/genetics.115.181453 Lewontin RC, Krakauer J (1973) Distribution of gene frequency as a test of the theory of selective neutrality of polymorphisms. Genetics 74: 175–195. https://doi.org/10.1093/genetics/74.1.175 Smadja CM, Loire E, Caminade P, Severac D, Gautier M, Ganem G (2022) Divergence of olfactory receptors associated with the evolution of assortative mating and reproductive isolation in mice. bioRxiv, 2022.07.21.500634, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.07.21.500634 | Divergence of olfactory receptors associated with the evolution of assortative mating and reproductive isolation in mice | Carole M. Smadja, Etienne Loire, Pierre Caminade, Dany Severac, Mathieu Gautier, Guila Ganem | <p>Deciphering the genetic bases of behavioural traits is essential to understanding how they evolve and contribute to adaptation and biological diversification, but it remains a substantial challenge, especially for behavioural traits with polyge... | | Adaptation, Behavior & Social Evolution, Genotype-Phenotype, Speciation | Christelle Fraïsse | 2022-07-25 11:54:52 | ||
24 Mar 2023
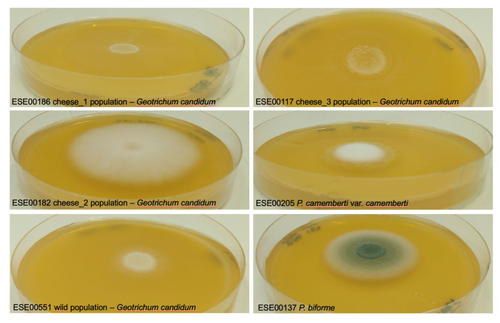
Domestication of different varieties in the cheese-making fungus Geotrichum candidumDiverse outcomes in cheese fungi domesticationRecommended by Christelle Fraïsse based on reviews by Delphine Sicard and 1 anonymous reviewer
Domestication is a complex process that imprints the demography and the genomes of domesticated populations, enforcing strong selective pressures on traits favourable to humans, e.g. for food production [1]. Domestication has been quite intensely studied in plants and animals, but less so in micro-organisms such as fungi, despite their assets (e.g. their small genomes and tractability in the lab). This elegant study by Bennetot and collaborators [2] on the cheese-making fungus Geotrichum candidum adds to the mounting body of studies in the genomics of fungi, proving they are excellent models in evolutionary biology for studying adaptation and drift in eukaryotes [3]. Bennetot et al. newly showed with whole genome sequences that all G. candidum strains isolated from cheese form a monophyletic clade subdivided into three genetically differentiated populations with several admixed strains, while the wild strains sampled from diverse geographic locations form a sister clade. This suggests the wild progenitor was not sampled in the present study and calls for future exciting work on the domestication history of the G. candidum fungus. The authors scanned the genomes for footprints of adaptation to the cheese environment and identified promising candidates, such as a gene involved in iron uptake (this element is limiting in cheese). Their functional genome analysis also provides evidence for higher contents of transposable elements in cheese-making strains, likely due to relaxed selection during the domestication process. This paper is particularly impressive in that the authors complemented the population genomic approach with the phenotypic characterization of the strains and tested their ability to outcompete common fungal food spoilers. The authors convincingly showed that cheese-making strains display phenotypic differences relative to wild relatives for multiple traits such as slower growth, lower proteolysis activity and a greater amount of volatiles attractive to consumers, these phenotypes being beneficial for cheese making. Finally, this work is particularly inspiring because it thoroughly discusses convergent evolution during domestication in different cheese-associated fungi. Indeed, studying populations experiencing similar environmental pressures is fundamental to understanding whether evolution is repeatable [4]. For instance, all three cheese populations of G. candidum exhibit a lower genetic diversity than wild populations. However, only one population displays a stronger domestication syndrome, resembling the Penicillium camemberti situation [5]. Furthermore, different cheese-making practices may have led to varying situations with clonal lineages in non-Roquefort P. roqueforti and P. camemberti [5, 6], while the cheese-making G. candidum populations still harbour some diversity. In a nutshell, Bennetot's study makes an important contribution to evolutionary biology and highlights the value of diversifying our model organisms toward under-represented clades. REFERENCES [1] Diamond J (2002) Evolution, consequences and future of plant and animal domestication. Nature 418: 700–707. https://doi.org/10.1038/nature01019 [2] Bennetot B, Vernadet J-P, Perkins V, Hautefeuille S, Rodríguez de la Vega RC, O’Donnell S, Snirc A, Grondin C, Lessard M-H, Peron A-C, Labrie S, Landaud S, Giraud T, Ropars J (2023) Domestication of different varieties in the cheese-making fungus Geotrichum candidum. bioRxiv, 2022.05.17.492043, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.05.17.492043 [3] Gladieux P, Ropars J, Badouin H, Branca A, Aguileta G, de Vienne DM, Rodríguez de la Vega RC, Branco S, Giraud T (2014) Fungal evolutionary genomics provides insight into the mechanisms of adaptive divergence in eukaryotes. Mol. Ecol. 23: 753–773. https://doi.org/10.1111/mec.12631 [4] Bolnick DI, Barrett RD, Oke KB, Rennison DJ, Stuart YE (2018) (Non)Parallel evolution. Ann. Rev. Ecol. Evol. Syst. 49: 303–330. https://doi.org/10.1146/annurev-ecolsys-110617-062240 [5] Ropars J, Didiot E, Rodríguez de la Vega RC, Bennetot B, Coton M, Poirier E, Coton E, Snirc A, Le Prieur S, Giraud T (2020) Domestication of the Emblematic White Cheese-Making Fungus Penicillium camemberti and Its Diversification into Two Varieties. Current Biol. 30: 4441–4453.e4. https://doi.org/10.1016/j.cub.2020.08.082 [6] Dumas, E, Feurtey, A, Rodríguez de la Vega, RC, Le Prieur S, Snirc A, Coton M, Thierry A, Coton E, Le Piver M, Roueyre D, Ropars J, Branca A, Giraud T (2020) Independent domestication events in the blue-cheese fungus Penicillium roqueforti. Mol Ecol. 29: 2639–2660. https://doi.org/10.1111/mec.15359 | Domestication of different varieties in the cheese-making fungus *Geotrichum candidum* | Bastien Bennetot, Jean-Philippe Vernadet, Vincent Perkins, Sophie Hautefeuille, Ricardo C. Rodríguez de la Vega, Samuel O’Donnell, Alodie Snirc, Cécile Grondin, Marie-Hélène Lessard, Anne-Claire Peron, Steve Labrie, Sophie Landaud, Tatiana Giraud,... | <p>Domestication is an excellent model for studying adaptation processes, involving recent adaptation and diversification, convergence following adaptation to similar conditions, as well as degeneration of unused functions. <em>Geotrichum candidum... | | Adaptation, Genome Evolution, Population Genetics / Genomics | Christelle Fraïsse | 2022-08-12 20:50:42 | ||
23 Jun 2021

Evolution of flowering time in a selfing annual plant: Roles of adaptation and genetic driftSeparating adaptation from drift: A cautionary tale from a self-fertilizing plantRecommended by Christoph Haag based on reviews by Pierre Olivier Cheptou, Jon Agren and Stefan LaurentIn recent years many studies have documented shifts in phenology in response to climate change, be it in arrival times in migrating birds, budset in trees, adult emergence in butterflies, or flowering time in annual plants (Coen et al. 2018; Piao et al. 2019). While these changes are, in part, explained by phenotypic plasticity, more and more studies find that they involve also genetic changes, that is, they involve evolutionary change (e.g., Metz et al. 2020). Yet, evolutionary change may occur through genetic drift as well as selection. Therefore, in order to demonstrate adaptive evolutionary change in response to climate change, drift has to be excluded as an alternative explanation (Hansen et al. 2012). A new study by Gay et al. (2021) shows just how difficult this can be. The authors investigated a recent evolutionary shift in flowering time by in a population an annual plant that reproduces predominantly by self-fertilization. The population has recently been subjected to increased temperatures and reduced rainfalls both of which are believed to select for earlier flowering times. They used a “resurrection” approach (Orsini et al. 2013; Weider et al. 2018): Genotypes from the past (resurrected from seeds) were compared alongside more recent genotypes (from more recently collected seeds) under identical conditions in the greenhouse. Using an experimental design that replicated genotypes, eliminated maternal effects, and controlled for microenvironmental variation, they found said genetic change in flowering times: Genotypes obtained from recently collected seeds flowered significantly (about 2 days) earlier than those obtained 22 generations before. However, neutral markers (microsatellites) also showed strong changes in allele frequencies across the 22 generations, suggesting that effective population size, Ne, was low (i.e., genetic drift was strong), which is typical for highly self-fertilizing populations. In addition, several multilocus genotypes were present at high frequencies and persisted over the 22 generations, almost as in clonal populations (e.g., Schaffner et al. 2019). The challenge was thus to evaluate whether the observed evolutionary change was the result of an adaptive response to selection or may be explained by drift alone. Here, Gay et al. (2021) took a particularly careful and thorough approach. First, they carried out a selection gradient analysis, finding that earlier-flowering plants produced more seeds than later-flowering plants. This suggests that, under greenhouse conditions, there was indeed selection for earlier flowering times. Second, investigating other populations from the same region (all populations are located on the Mediterranean island of Corsica, France), they found that a concurrent shift to earlier flowering times occurred also in these populations. Under the hypothesis that the populations can be regarded as independent replicates of the evolutionary process, the observation of concurrent shifts rules out genetic drift (under drift, the direction of change is expected to be random). The study may well have stopped here, concluding that there is good evidence for an adaptive response to selection for earlier flowering times in these self-fertilizing plants, at least under the hypothesis that selection gradients estimated in the greenhouse are relevant to field conditions. However, the authors went one step further. They used the change in the frequencies of the multilocus genotypes across the 22 generations as an estimate of realized fitness in the field and compared them to the phenotypic assays from the greenhouse. The results showed a tendency for high-fitness genotypes (positive frequency changes) to flower earlier and to produce more seeds than low-fitness genotypes. However, a simulation model showed that the observed correlations could be explained by drift alone, as long as Ne is lower than ca. 150 individuals. The findings were thus consistent with an adaptive evolutionary change in response to selection, but drift could only be excluded as the sole explanation if the effective population size was large enough. The study did provide two estimates of Ne (19 and 136 individuals, based on individual microsatellite loci or multilocus genotypes, respectively), but both are problematic. First, frequency changes over time may be influenced by the presence of a seed bank or by immigration from a genetically dissimilar population, which may lead to an underestimation of Ne (Wang and Whitlock 2003). Indeed, the low effective size inferred from the allele frequency changes at microsatellite loci appears to be inconsistent with levels of genetic diversity found in the population. Moreover, high self-fertilization reduces effective recombination and therefore leads to non-independence among loci. This lowers the precision of the Ne estimates (due to a higher sampling variance) and may also violate the assumption of neutrality due to the possibility of selection (e.g., due to inbreeding depression) at linked loci, which may be anywhere in the genome in case of high degrees of self-fertilization. There is thus no definite answer to the question of whether or not the observed changes in flowering time in this population were driven by selection. The study sets high standards for other, similar ones, in terms of thoroughness of the analyses and care in interpreting the findings. It also serves as a very instructive reminder to carefully check the assumptions when estimating neutral expectations, especially when working on species with complicated demographies or non-standard life cycles. Indeed the issues encountered here, in particular the difficulty of establishing neutral expectations in species with low effective recombination, may apply to many other species, including partially or fully asexual ones (Hartfield 2016). Furthermore, they may not be limited to estimating Ne but may also apply, for instance, to the establishment of neutral baselines for outlier analyses in genome scans (see e.g, Orsini et al. 2012). References Cohen JM, Lajeunesse MJ, Rohr JR (2018) A global synthesis of animal phenological responses to climate change. Nature Climate Change, 8, 224–228. https://doi.org/10.1038/s41558-018-0067-3 Gay L, Dhinaut J, Jullien M, Vitalis R, Navascués M, Ranwez V, Ronfort J (2021) Evolution of flowering time in a selfing annual plant: Roles of adaptation and genetic drift. bioRxiv, 2020.08.21.261230, ver. 4 recommended and peer-reviewed by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.08.21.261230 Hansen MM, Olivieri I, Waller DM, Nielsen EE (2012) Monitoring adaptive genetic responses to environmental change. Molecular Ecology, 21, 1311–1329. https://doi.org/10.1111/j.1365-294X.2011.05463.x Hartfield M (2016) Evolutionary genetic consequences of facultative sex and outcrossing. Journal of Evolutionary Biology, 29, 5–22. https://doi.org/10.1111/jeb.12770 Metz J, Lampei C, Bäumler L, Bocherens H, Dittberner H, Henneberg L, Meaux J de, Tielbörger K (2020) Rapid adaptive evolution to drought in a subset of plant traits in a large-scale climate change experiment. Ecology Letters, 23, 1643–1653. https://doi.org/10.1111/ele.13596 Orsini L, Schwenk K, De Meester L, Colbourne JK, Pfrender ME, Weider LJ (2013) The evolutionary time machine: using dormant propagules to forecast how populations can adapt to changing environments. Trends in Ecology & Evolution, 28, 274–282. https://doi.org/10.1016/j.tree.2013.01.009 Orsini L, Spanier KI, Meester LD (2012) Genomic signature of natural and anthropogenic stress in wild populations of the waterflea Daphnia magna: validation in space, time and experimental evolution. Molecular Ecology, 21, 2160–2175. https://doi.org/10.1111/j.1365-294X.2011.05429.x Piao S, Liu Q, Chen A, Janssens IA, Fu Y, Dai J, Liu L, Lian X, Shen M, Zhu X (2019) Plant phenology and global climate change: Current progresses and challenges. Global Change Biology, 25, 1922–1940. https://doi.org/10.1111/gcb.14619 Schaffner LR, Govaert L, De Meester L, Ellner SP, Fairchild E, Miner BE, Rudstam LG, Spaak P, Hairston NG (2019) Consumer-resource dynamics is an eco-evolutionary process in a natural plankton community. Nature Ecology & Evolution, 3, 1351–1358. https://doi.org/10.1038/s41559-019-0960-9 Wang J, Whitlock MC (2003) Estimating Effective Population Size and Migration Rates From Genetic Samples Over Space and Time. Genetics, 163, 429–446. PMID: 12586728 Weider LJ, Jeyasingh PD, Frisch D (2018) Evolutionary aspects of resurrection ecology: Progress, scope, and applications—An overview. Evolutionary Applications, 11, 3–10. https://doi.org/10.1111/eva.12563 | Evolution of flowering time in a selfing annual plant: Roles of adaptation and genetic drift | Laurène Gay, Julien Dhinaut, Margaux Jullien, Renaud Vitalis, Miguel Navascués, Vincent Ranwez, and Joëlle Ronfort | <p style="text-align: justify;">Resurrection studies are a useful tool to measure how phenotypic traits have changed in populations through time. If these traits modifications correlate with the environmental changes that occurred during the time ... | | Adaptation, Evolutionary Ecology, Genotype-Phenotype, Phenotypic Plasticity, Population Genetics / Genomics, Quantitative Genetics, Reproduction and Sex | Christoph Haag | 2020-08-21 17:26:59 | ||
17 Jun 2022

Spontaneous parthenogenesis in the parasitoid wasp Cotesia typhae: low frequency anomaly or evolving process?The potential evolutionary importance of low-frequency flexibility in reproductive modesRecommended by Christoph Haag based on reviews by Michael Lattorff and Jens BastOccasional events of asexual reproduction in otherwise sexual taxa have been documented since a long time. Accounts range from observations of offspring development from unfertilized eggs in Drosophila to rare offspring production by isolated females in lizards and birds (e.g., Stalker 1954, Watts et al 2006, Ryder et al. 2021). Many more such cases likely await documentation, as rare events are inherently difficult to observe. These rare events of asexual reproduction are often associated with low offspring fitness (“tychoparthenogenesis”), and have mostly been discarded in the evolutionary literature as reproductive accidents without evolutionary significance. Recently, however, there has been an increased interest in the details of evolutionary transitions from sexual to asexual reproduction (e.g., Archetti 2010, Neiman et al.2014, Lenormand et al. 2016), because these details may be key to understanding why successful transitions are rare, why they occur more frequently in some groups than in others, and why certain genetic mechanisms of ploidy maintenance or ploidy restoration are more often observed than others. In this context, the hypothesis has been formulated that regular or even obligate asexual reproduction may evolve from these rare events of asexual reproduction (e.g., Schwander et al. 2010). A new study by Capdevielle Dulac et al. (2022) now investigates this question in a parasitoid wasp, highlighting also the fact that what is considered rare or occasional may differ from one system to the next. The results show “rare” parthenogenetic production of diploid daughters occurring at variable frequencies (from zero to 2 %) in different laboratory strains, as well as in a natural population. They also demonstrate parthenogenetic production of female offspring in both virgin females and mated ones, as well as no reduced fecundity of parthenogenetically produced offspring. These findings suggest that parthenogenetic production of daughters, while still being rare, may be a more regular and less deleterious reproductive feature in this species than in other cases of occasional asexuality. Indeed, haplodiploid organisms, such as this parasitoid wasp have been hypothesized to facilitate evolutionary transitions to asexuality (Neimann et al. 2014, Van Der Kooi et al. 2017). First, in haploidiploid organisms, females are diploid and develop from normal, fertilized eggs, but males are haploid as they develop parthenogenetically from unfertilized eggs. This means that, in these species, fertilization is not necessarily needed to trigger development, thus removing one of the constraints for transitions to obligate asexuality (Engelstädter 2008, Vorburger 2014). Second, spermatogenesis in males occurs by a modified meiosis that skips the first meiotic division (e.g., Ferree et al. 2019). Haploidiploid organisms may thus have a potential route for an evolutionary transition to obligate parthenogenesis that is not available to organisms: The pathways for the modified meiosis may be re-used for oogenesis, which might result in unreduced, diploid eggs. Third, the particular species studied here regularly undergoes inbreeding by brother-sister mating within their hosts. Homozygosity, including at the sex determination locus (Engelstädter 2008), is therefore expected to have less negative effects in this species compared to many other, non-inbreeding haplodipoids (see also Little et al. 2017). This particular species may therefore be less affected by loss of heterozygosity, which occurs in a fashion similar to self-fertilization under many forms of non-clonal parthenogenesis. Indeed, the study also addresses the mechanisms underlying parthenogenesis in the species. Surprisingly, the authors find that parthenogenetically produced females are likely produced by two distinct genetic mechanisms. The first results in clonality (maintenance of the maternal genotype), whereas the second one results in a loss of heterozygosity towards the telomeres, likely due to crossovers occurring between the centromeres and the telomeres. Moreover, bacterial infections appear to affect the propensity of parthenogenesis but are unlikely the primary cause. Together, the finding suggests that parthenogenesis is a variable trait in the species, both in terms of frequency and mechanisms. It is not entirely clear to what degree this variation is heritable, but if it is, then these results constitute evidence for low-frequency existence of variable and heritable parthenogenesis phenotypes, that is, the raw material from which evolutionary transitions to more regular forms of parthenogenesis may occur.
References Archetti M (2010) Complementation, Genetic Conflict, and the Evolution of Sex and Recombination. Journal of Heredity, 101, S21–S33. https://doi.org/10.1093/jhered/esq009 Capdevielle Dulac C, Benoist R, Paquet S, Calatayud P-A, Obonyo J, Kaiser L, Mougel F (2022) Spontaneous parthenogenesis in the parasitoid wasp Cotesia typhae: low frequency anomaly or evolving process? bioRxiv, 2021.12.13.472356, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.12.13.472356 Engelstädter J (2008) Constraints on the evolution of asexual reproduction. BioEssays, 30, 1138–1150. https://doi.org/10.1002/bies.20833 Ferree PM, Aldrich JC, Jing XA, Norwood CT, Van Schaick MR, Cheema MS, Ausió J, Gowen BE (2019) Spermatogenesis in haploid males of the jewel wasp Nasonia vitripennis. Scientific Reports, 9, 12194. https://doi.org/10.1038/s41598-019-48332-9 van der Kooi CJ, Matthey-Doret C, Schwander T (2017) Evolution and comparative ecology of parthenogenesis in haplodiploid arthropods. Evolution Letters, 1, 304–316. https://doi.org/10.1002/evl3.30 Lenormand T, Engelstädter J, Johnston SE, Wijnker E, Haag CR (2016) Evolutionary mysteries in meiosis. Philosophical Transactions of the Royal Society B: Biological Sciences, 371, 20160001. https://doi.org/10.1098/rstb.2016.0001 Little CJ, Chapuis M-P, Blondin L, Chapuis E, Jourdan-Pineau H (2017) Exploring the relationship between tychoparthenogenesis and inbreeding depression in the Desert Locust, Schistocerca gregaria. Ecology and Evolution, 7, 6003–6011. https://doi.org/10.1002/ece3.3103 Neiman M, Sharbel TF, Schwander T (2014) Genetic causes of transitions from sexual reproduction to asexuality in plants and animals. Journal of Evolutionary Biology, 27, 1346–1359. https://doi.org/10.1111/jeb.12357 Ryder OA, Thomas S, Judson JM, Romanov MN, Dandekar S, Papp JC, Sidak-Loftis LC, Walker K, Stalis IH, Mace M, Steiner CC, Chemnick LG (2021) Facultative Parthenogenesis in California Condors. Journal of Heredity, 112, 569–574. https://doi.org/10.1093/jhered/esab052 Schwander T, Vuilleumier S, Dubman J, Crespi BJ (2010) Positive feedback in the transition from sexual reproduction to parthenogenesis. Proceedings of the Royal Society B: Biological Sciences, 277, 1435–1442. https://doi.org/10.1098/rspb.2009.2113 Stalker HD (1954) Parthenogenesis in Drosophila. Genetics, 39, 4–34. https://doi.org/10.1093/genetics/39.1.4 Vorburger C (2014) Thelytoky and Sex Determination in the Hymenoptera: Mutual Constraints. Sexual Development, 8, 50–58. https://doi.org/10.1159/000356508 Watts PC, Buley KR, Sanderson S, Boardman W, Ciofi C, Gibson R (2006) Parthenogenesis in Komodo dragons. Nature, 444, 1021–1022. https://doi.org/10.1038/4441021a | Spontaneous parthenogenesis in the parasitoid wasp Cotesia typhae: low frequency anomaly or evolving process? | Claire Capdevielle Dulac, Romain Benoist, Sarah Paquet, Paul-André Calatayud, Julius Obonyo, Laure Kaiser, Florence Mougel | <p style="text-align: justify;">Hymenopterans are haplodiploids and unlike most other Arthropods they do not possess sexual chromosomes. Sex determination typically happens via the ploidy of individuals: haploids become males and diploids become f... | | Evolutionary Ecology, Life History, Reproduction and Sex | Christoph Haag | 2021-12-16 15:25:16 | ||
03 May 2020

When does gene flow facilitate evolutionary rescue?Reconciling the upsides and downsides of migration for evolutionary rescueRecommended by Claudia Bank based on reviews by 3 anonymous reviewersThe evolutionary response of populations to changing or novel environments is a topic that unites the interests of evolutionary biologists, ecologists, and biomedical researchers [1]. A prominent phenomenon in this research area is evolutionary rescue, whereby a population that is otherwise doomed to extinction survives due to the spread of new or pre-existing mutations that are beneficial in the new environment. Scenarios of evolutionary rescue require a specific set of parameters: the absolute growth rate has to be negative before the rescue mechanism spreads, upon which the growth rate becomes positive. However, potential examples of its relevance exist (e.g., [2]). From a theoretical point of view, the technical challenge but also the beauty of evolutionary rescue models is that they combine the study of population dynamics (i.e., changes in the size of populations) and population genetics (i.e., changes in the frequencies in the population). Together, the potential relevance of evolutionary rescue in nature and the models' theoretical appeal has resulted in a suite of modeling studies on the subject in recent years. References [1] Bell, G. (2017). Evolutionary Rescue. Annual Review of Ecology, Evolution, and Systematics 48(1), 605-627. doi: 10.1146/annurev-ecolsys-110316-023011 | When does gene flow facilitate evolutionary rescue? | Matteo Tomasini, Stephan Peischl | <p>Experimental and theoretical studies have highlighted the impact of gene flow on the probability of evolutionary rescue in structured habitats. Mathematical modelling and simulations of evolutionary rescue in spatially or otherwise structured p... | | Evolutionary Dynamics, Evolutionary Theory, Population Genetics / Genomics | Claudia Bank | 2019-05-22 11:12:13 | ||
16 Dec 2016
POSTPRINT
Spatiotemporal microbial evolution on antibiotic landscapesA poster child for experimental evolutionRecommended by Daniel Rozen and Arjan de VisserEvolution is usually studied via two distinct approaches: by inferring evolutionary processes from relatedness patterns among living species or by observing evolution in action in the laboratory or field. A recent study by Baym and colleagues in Science [1] has now combined these approaches by taking advantage of the pattern left behind by spatially evolving bacterial populations. Evolution is often considered too slow to see, and can only be inferred by studying patterns of relatedness using phylogenetic trees. Increasingly, however, researchers are moving nature into the lab and watching as evolution unfolds under their noses. The field of experimental evolution follows evolutionary change in the laboratory over 10s to 1000s of generations, yielding insights into bacterial, viral, plant, or fly evolution (among many other species) that are simply not possible in the field. Yet, as powerful as experimental evolution is, it lacks a posterchild. There is no Galapagos finch radiation, nor a stunning series of cichlids to showcase to our students to pique their interests. Let’s face it, E. coli is no stickleback! And while practitioners of experimental evolution can explain the virtues of examining 60,000 generations of bacterial evolution in action, appreciating this nevertheless requires a level of insight and imagination that often eludes students, who need to see “it” to get it. Enter MEGA, an idea and a film that could become the new face of experimental evolution. It replaces big numbers of generations or images of scientists, with an actual picture of the scientific result. MEGA, or Microbial Evolution and Growth Arena, is essentially an enormous petri dish and is the brainchild of Michael Baym, Tami Leiberman and their colleagues in Roy Kishony’s lab at Technion Israel Institute of Technology and Harvard Medical School. The idea of MEGA is to allow bacteria to swim over a spatially defined landscape while adapting to the local conditions, in this case antibiotics. When bacteria are inoculated onto one end of the plate they consume resources while swarming forward from the plate edge. In a few days, the bacteria grow into an area with antibiotics to which they are susceptible. This stops growth until a mutation arises that permits the bacteria to jump this hurdle, after which growth proceeds until the next hurdle of a 10-fold higher antibiotic concentration, and so on. By this simple approach, Baym et al. [1] evolved E. coli that were nearly 105-fold more resistant to two different antibiotics in just over 10 days. In addition, they identified the mutations that were required for these changes, showed that mutations conferring smaller benefits were required before bacteria could evolve maximal resistance, observed changes to the mutation rate, and demonstrated the importance of spatial structure in constraining adaptation. For one thing, the rate of resistance evolution is impressive, and also quite scary given the mounting threat of antibiotic-resistant pathogens. However, MEGA also offers a uniquely visual insight into evolutionary change. By taking successive images of the MEGA plate, the group was able to watch the bacteria move, get trapped because of their susceptibility to the antibiotic, and then get past these traps as new mutations emerged that increased resistance. Each transition showcases evolution in real time. In addition, by leaving a spatial pattern of evolutionary steps behind, the MEGA plate offers unique opportunities to thoroughly investigate these steps when the experiment is finished. For instance, subsequent steps in mutational pathways can be characterized, but also their effects on fitness can be quantified in situ by measuring changes in survival and reproduction. This new method is undoubtedly a boon to the field of experimental evolution and offers endless opportunities for experimental elaboration. Perhaps of equal importance, MEGA is a tool that is portable to the classroom and to the public at large. Don’t believe in evolution? Watch this. You only have time for a short internship or lab practical? No problem. Don’t worry much about antibiotic resistance? Check this out. Like the best experimental tools, MEGA is simple but allows for complicated insights. And even if it is less charismatic than a finch, it still allows for the kinds of “gee-whiz” insights that will get students hooked on evolutionary biology. Reference [1] Baym M, Lieberman TD, Kelsic ED, Chait R, Gross R, Yelin I, Kishony R. 2016. Spatiotemporal microbial evolution on antibiotic landscapes. Science 353:1147-1151. doi: 10.1126/science.aag0822 | Spatiotemporal microbial evolution on antibiotic landscapes | Baym M, Lieberman TD, Kelsic ED, Chait R, Gross R, Yelin I, Kishony R | A key aspect of bacterial survival is the ability to evolve while migrating across spatially varying environmental challenges. Laboratory experiments, however, often study evolution in well-mixed systems. Here, we introduce an experimental device,... | | Adaptation, Evolutionary Applications, Experimental Evolution | Daniel Rozen | 2016-12-14 14:26:06 | ||
11 Dec 2020

Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence dataPhylodynamics of hepatitis C virus reveals transmission dynamics within and between risk groups in LyonRecommended by David Rasmussen based on reviews by Chris Wymant and Louis DuPlessisGenomic epidemiology seeks to better understand the transmission dynamics of infectious pathogens using molecular sequence data. Phylodynamic methods have given genomic epidemiology new power to track the transmission dynamics of pathogens by combining phylogenetic analyses with epidemiological modeling. In recent year, applications of phylodynamics to chronic viral infections such as HIV and hepatitis C virus (HVC) have provided some of the best examples of how phylodynamic inference can provide valuable insights into transmission dynamics within and between different subpopulations or risk groups, allowing for more targeted interventions. References [1] Rasmussen, D. A., Volz, E. M., and Koelle, K. (2014). Phylodynamic inference for structured epidemiological models. PLoS Comput Biol, 10(4), e1003570. doi: https://doi.org/10.1371/journal.pcbi.1003570 | Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence data | Gonche Danesh, Victor Virlogeux, Christophe Ramière, Caroline Charre, Laurent Cotte, Samuel Alizon | <p>Opioid substitution and syringes exchange programs have drastically reduced hepatitis C virus (HCV) spread in France but HCV sexual transmission in men having sex with men (MSM) has recently arisen as a significant public health concern. The fa... | | Evolutionary Epidemiology, Phylogenetics / Phylogenomics | David Rasmussen | 2019-07-11 13:37:23 | ||
13 Dec 2016
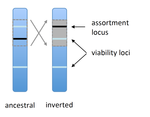
POSTPRINT
Prezygotic isolation, mating preferences, and the evolution of chromosomal inversionsThe spread of chromosomal inversions as a mechanism for reinforcementRecommended by Denis Roze and Thomas Broquet
Several examples of chromosomal inversions carrying genes affecting mate choice have been reported from various organisms. Furthermore, inversions are also frequently involved in genetic isolation between populations or species. Past work has shown that inversions can spread when they capture not only some loci involved in mate choice but also loci involved in incompatibilities between hybridizing populations [1]. In this new paper [2], the authors derive analytical approximations for the selection coefficient associated with an inversion suppressing recombination between a locus involved in mate choice and one (or several) locus involved in Dobzhansky-Muller incompatibilities. Two mechanisms for mate choice are considered: assortative mating based on the allele present at a single locus, or a trait-preference model where one locus codes for the trait and another for the preference. The results show that such an inversion is generally favoured, the selective advantage associated with the inversion being strongest when hybridization is sufficiently frequent. Assuming pairwise epistatic interactions between loci involved in incompatibilities, selection for the inversion increases approximately linearly with the number of such loci captured by the inversion. This paper is a good read for several reasons. First, it presents the problem clearly (e.g. the introduction provides a clear and concise presentation of the issue and past work) and its crystal-clear writing facilitates the reader's understanding of theoretical approaches and results. Second, the analysis is competently done and adds to previous work by showing that very general conditions are expected to be favourable to the spread of the type of inversion considered here. And third, it provides food for thought about the role of inversions in the origin or the reinforcement of divergence between nascent species. One result of this work is that an inversion linked to pre-zygotic isolation "is favoured so long as there is viability selection against recombinant genotypes", suggesting that genetic incompatibilities must have evolved first and that inversions capturing mating preference loci may then enhance pre-existing reproductive isolation. However, the results also show that inversions are more likely to be favoured in hybridizing populations among which gene flow is still high, rather than in more strongly isolated populations. This matches the observation that inversions are more frequently observed between sympatric species than between allopatric ones. References [1] Trickett AJ, Butlin RK. 1994. Recombination Suppressors and the Evolution of New Species. Heredity 73:339-345. doi: 10.1038/hdy.1994.180 [2] Dagilis AJ, Kirkpatrick M. 2016. Prezygotic isolation, mating preferences, and the evolution of chromosomal inversions. Evolution 70: 1465–1472. doi: 10.1111/evo.12954 | Prezygotic isolation, mating preferences, and the evolution of chromosomal inversions | Dagilis AJ, Kirkpatrick M | Chromosomal inversions are frequently implicated in isolating species. Models have shown how inversions can evolve in the context of postmating isolation. Inversions are also frequently associated with mating preferences, a topic that has not been... | | Adaptation, Evolutionary Theory, Genome Evolution, Hybridization / Introgression, Population Genetics / Genomics, Speciation | Denis Roze | 2016-12-13 22:11:54 |




