
Latest recommendations

| Id | Title | Authors | Abstract | Picture▲ | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
17 Jun 2022

Spontaneous parthenogenesis in the parasitoid wasp Cotesia typhae: low frequency anomaly or evolving process?The potential evolutionary importance of low-frequency flexibility in reproductive modesRecommended by Christoph Haag based on reviews by Michael Lattorff and Jens BastOccasional events of asexual reproduction in otherwise sexual taxa have been documented since a long time. Accounts range from observations of offspring development from unfertilized eggs in Drosophila to rare offspring production by isolated females in lizards and birds (e.g., Stalker 1954, Watts et al 2006, Ryder et al. 2021). Many more such cases likely await documentation, as rare events are inherently difficult to observe. These rare events of asexual reproduction are often associated with low offspring fitness (“tychoparthenogenesis”), and have mostly been discarded in the evolutionary literature as reproductive accidents without evolutionary significance. Recently, however, there has been an increased interest in the details of evolutionary transitions from sexual to asexual reproduction (e.g., Archetti 2010, Neiman et al.2014, Lenormand et al. 2016), because these details may be key to understanding why successful transitions are rare, why they occur more frequently in some groups than in others, and why certain genetic mechanisms of ploidy maintenance or ploidy restoration are more often observed than others. In this context, the hypothesis has been formulated that regular or even obligate asexual reproduction may evolve from these rare events of asexual reproduction (e.g., Schwander et al. 2010). A new study by Capdevielle Dulac et al. (2022) now investigates this question in a parasitoid wasp, highlighting also the fact that what is considered rare or occasional may differ from one system to the next. The results show “rare” parthenogenetic production of diploid daughters occurring at variable frequencies (from zero to 2 %) in different laboratory strains, as well as in a natural population. They also demonstrate parthenogenetic production of female offspring in both virgin females and mated ones, as well as no reduced fecundity of parthenogenetically produced offspring. These findings suggest that parthenogenetic production of daughters, while still being rare, may be a more regular and less deleterious reproductive feature in this species than in other cases of occasional asexuality. Indeed, haplodiploid organisms, such as this parasitoid wasp have been hypothesized to facilitate evolutionary transitions to asexuality (Neimann et al. 2014, Van Der Kooi et al. 2017). First, in haploidiploid organisms, females are diploid and develop from normal, fertilized eggs, but males are haploid as they develop parthenogenetically from unfertilized eggs. This means that, in these species, fertilization is not necessarily needed to trigger development, thus removing one of the constraints for transitions to obligate asexuality (Engelstädter 2008, Vorburger 2014). Second, spermatogenesis in males occurs by a modified meiosis that skips the first meiotic division (e.g., Ferree et al. 2019). Haploidiploid organisms may thus have a potential route for an evolutionary transition to obligate parthenogenesis that is not available to organisms: The pathways for the modified meiosis may be re-used for oogenesis, which might result in unreduced, diploid eggs. Third, the particular species studied here regularly undergoes inbreeding by brother-sister mating within their hosts. Homozygosity, including at the sex determination locus (Engelstädter 2008), is therefore expected to have less negative effects in this species compared to many other, non-inbreeding haplodipoids (see also Little et al. 2017). This particular species may therefore be less affected by loss of heterozygosity, which occurs in a fashion similar to self-fertilization under many forms of non-clonal parthenogenesis. Indeed, the study also addresses the mechanisms underlying parthenogenesis in the species. Surprisingly, the authors find that parthenogenetically produced females are likely produced by two distinct genetic mechanisms. The first results in clonality (maintenance of the maternal genotype), whereas the second one results in a loss of heterozygosity towards the telomeres, likely due to crossovers occurring between the centromeres and the telomeres. Moreover, bacterial infections appear to affect the propensity of parthenogenesis but are unlikely the primary cause. Together, the finding suggests that parthenogenesis is a variable trait in the species, both in terms of frequency and mechanisms. It is not entirely clear to what degree this variation is heritable, but if it is, then these results constitute evidence for low-frequency existence of variable and heritable parthenogenesis phenotypes, that is, the raw material from which evolutionary transitions to more regular forms of parthenogenesis may occur.
References Archetti M (2010) Complementation, Genetic Conflict, and the Evolution of Sex and Recombination. Journal of Heredity, 101, S21–S33. https://doi.org/10.1093/jhered/esq009 Capdevielle Dulac C, Benoist R, Paquet S, Calatayud P-A, Obonyo J, Kaiser L, Mougel F (2022) Spontaneous parthenogenesis in the parasitoid wasp Cotesia typhae: low frequency anomaly or evolving process? bioRxiv, 2021.12.13.472356, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.12.13.472356 Engelstädter J (2008) Constraints on the evolution of asexual reproduction. BioEssays, 30, 1138–1150. https://doi.org/10.1002/bies.20833 Ferree PM, Aldrich JC, Jing XA, Norwood CT, Van Schaick MR, Cheema MS, Ausió J, Gowen BE (2019) Spermatogenesis in haploid males of the jewel wasp Nasonia vitripennis. Scientific Reports, 9, 12194. https://doi.org/10.1038/s41598-019-48332-9 van der Kooi CJ, Matthey-Doret C, Schwander T (2017) Evolution and comparative ecology of parthenogenesis in haplodiploid arthropods. Evolution Letters, 1, 304–316. https://doi.org/10.1002/evl3.30 Lenormand T, Engelstädter J, Johnston SE, Wijnker E, Haag CR (2016) Evolutionary mysteries in meiosis. Philosophical Transactions of the Royal Society B: Biological Sciences, 371, 20160001. https://doi.org/10.1098/rstb.2016.0001 Little CJ, Chapuis M-P, Blondin L, Chapuis E, Jourdan-Pineau H (2017) Exploring the relationship between tychoparthenogenesis and inbreeding depression in the Desert Locust, Schistocerca gregaria. Ecology and Evolution, 7, 6003–6011. https://doi.org/10.1002/ece3.3103 Neiman M, Sharbel TF, Schwander T (2014) Genetic causes of transitions from sexual reproduction to asexuality in plants and animals. Journal of Evolutionary Biology, 27, 1346–1359. https://doi.org/10.1111/jeb.12357 Ryder OA, Thomas S, Judson JM, Romanov MN, Dandekar S, Papp JC, Sidak-Loftis LC, Walker K, Stalis IH, Mace M, Steiner CC, Chemnick LG (2021) Facultative Parthenogenesis in California Condors. Journal of Heredity, 112, 569–574. https://doi.org/10.1093/jhered/esab052 Schwander T, Vuilleumier S, Dubman J, Crespi BJ (2010) Positive feedback in the transition from sexual reproduction to parthenogenesis. Proceedings of the Royal Society B: Biological Sciences, 277, 1435–1442. https://doi.org/10.1098/rspb.2009.2113 Stalker HD (1954) Parthenogenesis in Drosophila. Genetics, 39, 4–34. https://doi.org/10.1093/genetics/39.1.4 Vorburger C (2014) Thelytoky and Sex Determination in the Hymenoptera: Mutual Constraints. Sexual Development, 8, 50–58. https://doi.org/10.1159/000356508 Watts PC, Buley KR, Sanderson S, Boardman W, Ciofi C, Gibson R (2006) Parthenogenesis in Komodo dragons. Nature, 444, 1021–1022. https://doi.org/10.1038/4441021a | Spontaneous parthenogenesis in the parasitoid wasp Cotesia typhae: low frequency anomaly or evolving process? | Claire Capdevielle Dulac, Romain Benoist, Sarah Paquet, Paul-André Calatayud, Julius Obonyo, Laure Kaiser, Florence Mougel | <p style="text-align: justify;">Hymenopterans are haplodiploids and unlike most other Arthropods they do not possess sexual chromosomes. Sex determination typically happens via the ploidy of individuals: haploids become males and diploids become f... | | Evolutionary Ecology, Life History, Reproduction and Sex | Christoph Haag | 2021-12-16 15:25:16 | ||
04 Mar 2021
Simulation of bacterial populations with SLiMSimulating bacterial evolution forward-in-timeRecommended by Frederic Bertels based on reviews by 3 anonymous reviewersJean Cury and colleagues (2021) have developed a protocol to simulate bacterial evolution in SLiM. In contrast to existing methods that depend on the coalescent, SLiM simulates evolution forward in time. SLiM has, up to now, mostly been used to simulate the evolution of eukaryotes (Haller and Messer 2019), but has been adapted here to simulate evolution in bacteria. Forward-in-time simulations are usually computationally very costly. To circumvent this issue, bacterial population sizes are scaled down. One would now expect results to become inaccurate, however, Cury et al. show that scaled-down forwards simulations provide very accurate results (similar to those provided by coalescent simulators) that are consistent with theoretical expectations. Simulations were analyzed and compared to existing methods in simple and slightly more complex scenarios where recombination affects evolution. In all scenarios, simulation results from coalescent methods (fastSimBac (De Maio and Wilson 2017), ms (Hudson 2002)) and scaled-down forwards simulations were very similar, which is very good news indeed. A biologist not aware of the complexities of forwards, backwards simulations and the coalescent, might now naïvely ask why another simulation method is needed if existing methods perform just as well. To address this question the manuscript closes with a very neat example of what exactly is possible with forwards simulations that cannot be achieved using existing methods. The situation modeled is the growth and evolution of a set of 50 bacteria that are randomly distributed on a petri dish. One side of the petri dish is covered in an antibiotic the other is antibiotic-free. Over time, the bacteria grow and acquire antibiotic resistance mutations until the entire artificial petri dish is covered with a bacterial lawn. This simulation demonstrates that it is possible to simulate extremely complex (e.g. real world) scenarios to, for example, assess whether certain phenomena are expected with our current understanding of bacterial evolution, or whether there are additional forces that need to be taken into account. Hence, forwards simulators could significantly help us to understand what current models can and cannot explain in evolutionary biology.
References Cury J, Haller BC, Achaz G, Jay F (2021) Simulation of bacterial populations with SLiM. bioRxiv, 2020.09.28.316869, version 5 peer-reviewed and recommended by Peer community in Evolutionary Biology. https://doi.org/10.1101/2020.09.28.316869 De Maio N, Wilson DJ (2017) The Bacterial Sequential Markov Coalescent. Genetics, 206, 333–343. https://doi.org/10.1534/genetics.116.198796 Haller BC, Messer PW (2019) SLiM 3: Forward Genetic Simulations Beyond the Wright–Fisher Model. Molecular Biology and Evolution, 36, 632–637. https://doi.org/10.1093/molbev/msy228 Hudson RR (2002) Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics, 18, 337–338. https://doi.org/10.1093/bioinformatics/18.2.337 | Simulation of bacterial populations with SLiM | Jean Cury, Benjamin C. Haller, Guillaume Achaz, and Flora Jay | <p>Simulation of genomic data is a key tool in population genetics, yet, to date, there is no forward-in-time simulator of bacterial populations that is both computationally efficient and adaptable to a wide range of scenarios. Here we demonstrate... | | Bioinformatics & Computational Biology, Population Genetics / Genomics | Frederic Bertels | 2020-10-02 19:03:42 | ||
01 Jul 2022

Genomic evidence of paternal genome elimination in the globular springtail Allacma fuscaPressing NGS data through the mill of Kmer spectra and allelic coverage ratios in order to scan reproductive modes in non-model speciesRecommended by Nicolas Bierne based on reviews by Paul Simion and 2 anonymous reviewersThe genomic revolution has given us access to inexpensive genetic data for any species. Simultaneously we have lost the ability to easily identify chimerism in samples or some unusual deviations from standard Mendelian genetics. Methods have been developed to identify sex chromosomes, characterise the ploidy, or understand the exact form of parthenogenesis from genomic data. However, we rarely consider that the tissues we extract DNA from could be a mixture of cells with different genotypes or karyotypes. This can nonetheless happen for a variety of (fascinating) reasons such as somatic chromosome elimination, transmissible cancer, or parental genome elimination. Without a dedicated analysis, it is very easy to miss it. In this preprint, Jaron et al. (2022) used an ingenious analysis of whole individual NGS data to test the hypothesis of paternal genome elimination in the globular springtail Allacma fusca. The authors suspected that a high fraction of the whole body of males is made of sperm in this species and if this species undergoes paternal genome elimination, we would expect that sperm would only contain maternally inherited chromosomes. Given the reference genome was highly fragmented, they developed a two-tissue model to analyse Kmer spectra and obtained confirmation that around one-third of the tissue was sperm in males. This allowed them to test whether coverage patterns were consistent with the species exhibiting paternal genome elimination. They combined their estimation of the fraction of haploid tissue with allele coverages in autosomes and the X chromosome to obtain support for a bias toward one parental allele, suggesting that all sperm carries the same parental haplotype. It could be the maternal or the paternal alleles, but paternal genome elimination is most compatible with the known biology of Arthropods. SNP calling was used to confirm conclusions based on the analysis of the raw pileups. I found this study to be a good example of how a clever analysis of Kmer spectra and allele coverages can provide information about unusual modes of reproduction in a species, even though it does not have a well-assembled genome yet. As advocated by the authors, routine inspection of Kmer spectra and allelic read-count distributions should be included in the best practice of NGS data analysis. They provide the method to identify paternal genome elimination but also the way to develop similar methods to detect another kind of genetic chimerism in the avalanche of sequence data produced nowadays. References Jaron KS, Hodson CN, Ellers J, Baird SJ, Ross L (2022) Genomic evidence of paternal genome elimination in the globular springtail Allacma fusca. bioRxiv, 2021.11.12.468426, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.11.12.468426 | Genomic evidence of paternal genome elimination in the globular springtail Allacma fusca | Kamil S. Jaron, Christina N. Hodson, Jacintha Ellers, Stuart JE Baird, Laura Ross | <p style="text-align: justify;">Paternal genome elimination (PGE) - a type of reproduction in which males inherit but fail to pass on their father’s genome - evolved independently in six to eight arthropod clades. Thousands of species, including s... | | Genome Evolution, Reproduction and Sex | Nicolas Bierne | 2021-11-18 00:09:43 | ||
03 Apr 2020

Evolution at two time-frames: ancient and common origin of two structural variants involved in local adaptation of the European plaice (Pleuronectes platessa)Genomic structural variants involved in local adaptation of the European plaiceRecommended by Maren Wellenreuther based on reviews by 3 anonymous reviewersAwareness has been growing that structural variants in the genome of species play a fundamental role in adaptive evolution and diversification [1]. Here, Le Moan and co-authors [2] report empirical genomic-wide SNP data on the European plaice (Pleuronectes platessa) across a major environmental transmission zone, ranging from the North Sea to the Baltic Sea. Regions of high linkage disequilibrium suggest the presence of two structural variants that appear to have evolved 220 kya. These two putative structural variants show weak signatures of isolation by distance when contrasted against the rest of the genome, but the frequency of the different putative structural variants appears to co-vary in some parts of the studied range with the environment, indicating the involvement of both selective and neutral processes. This study adds to the mounting body of evidence that structural genomic variants harbour significant information that allows species to respond and adapt to the local environmental context. References [1] Wellenreuther, M., Mérot, C., Berdan, E., & Bernatchez, L. (2019). Going beyond SNPs: the role of structural genomic variants in adaptive evolution and species diversification. Molecular ecology, 28(6), 1203-1209. doi: 10.1111/mec.15066 | Evolution at two time-frames: ancient and common origin of two structural variants involved in local adaptation of the European plaice (Pleuronectes platessa) | Alan Le Moan, Dorte Bekkevold & Jakob Hemmer-Hansen | <p>Changing environmental conditions can lead to population diversification through differential selection on standing genetic variation. Structural variant (SV) polymorphisms provide examples of ancient alleles that in time become associated with... | | Adaptation, Hybridization / Introgression, Population Genetics / Genomics, Speciation | Maren Wellenreuther | 2019-07-13 12:44:01 | ||
05 Aug 2020

Transposable Elements are an evolutionary force shaping genomic plasticity in the parthenogenetic root-knot nematode Meloidogyne incognitaDNA transposons drive genome evolution of the root-knot nematode Meloidogyne incognitaRecommended by Ines Alvarez based on reviews by Daniel Vitales and 2 anonymous reviewers based on reviews by Daniel Vitales and 2 anonymous reviewers
Duplications, mutations and recombination may be considered the main sources of genomic variation and evolution. In addition, sexual recombination is essential in purging deleterious mutations and allowing advantageous allelic combinations to occur (Glémin et al. 2019). However, in parthenogenetic asexual organisms, variation cannot be explained by sexual recombination, and other mechanisms must account for it. Although it is known that transposable elements (TE) may influence on genome structure and gene expression patterns, their role as a primary source of genomic variation and rapid adaptability has received less attention. An important role of TE on adaptive genome evolution has been documented for fungal phytopathogens (Faino et al. 2016), suggesting that TE activity might explain the evolutionary dynamics of this type of organisms. References [1] Bessereau J-L. 2006. Transposons in C. elegans. WormBook. 10.1895/wormbook.1.70.1 | Transposable Elements are an evolutionary force shaping genomic plasticity in the parthenogenetic root-knot nematode Meloidogyne incognita | Djampa KL Kozlowski, Rahim Hassanaly-Goulamhoussen, Martine Da Rocha, Georgios D Koutsovoulos, Marc Bailly-Bechet, Etienne GJ Danchin | <p>Despite reproducing without sexual recombination, the root-knot nematode Meloidogyne incognita is adaptive and versatile. Indeed, this species displays a global distribution, is able to parasitize a large range of plants and can overcome plant ... | | Adaptation, Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Population Genetics / Genomics, Reproduction and Sex | Ines Alvarez | 2020-05-04 11:43:14 | ||
02 Feb 2023
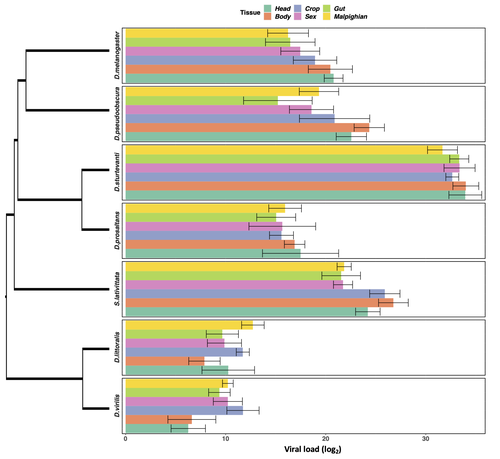
Heterogeneities in infection outcomes across species: sex and tissue differences in virus susceptibilitySusceptibility to infection is not explained by sex or differences in tissue tropism across different species of DrosophilaRecommended by Alison Duncan based on reviews by Greg Hurst and 1 anonymous reviewerUnderstanding factors explaining both intra and interspecific variation in susceptibility to infection by parasites remains a key question in evolutionary biology. Within a species variation in susceptibility is often explained by differences in behaviour affecting exposure to infection and/or resistance affecting the degree by which parasite growth is controlled (Roy & Kirchner, 2000, Behringer et al., 2000). This can vary between the sexes (Kelly et al., 2018) and may be explained by the ability of a parasite to attack different organs or tissues (Brierley et al., 2019). However, what goes on within one species is not always relevant to another, making it unclear when patterns can be scaled up and generalised across species. This is also important to understand when parasites may jump hosts, or identify species that may be susceptible to a host jump (Longdon et al., 2015). Phylogenetic distance between hosts is often an important factor explaining susceptibility to a particular parasite in plant and animal hosts (Gilbert & Webb, 2007, Faria et al., 2013). In two separate experiments, Roberts and Longdon (Roberts & Longdon, 2022) investigated how sex and tissue tropism affected variation in the load of Drosophila C Virus (DCV) across multiple Drosophila species. DCV load has been shown to correlate positively with mortality (Longdon et al., 2015). Overall, they found that load did not vary between the sexes; within a species males and females had similar DCV loads for 31 different species. There was some variation in levels of DCV growth in different tissue types, but these too were consistent across males for 7 species of Drosophila. Instead, in both experiments, host phylogeny or interspecific variation, explained differences in DCV load with some species being more infected than others. This study is neat in that it incorporates and explores simultaneously both intra and interspecific variation in infection-related life-history traits which is not often done (but see (Longdon et al., 2015, Imrie et al., 2021, Longdon et al., 2011, Johnson et al., 2012). Indeed, most studies to date explore either inter-specific differences in susceptibility to a parasite (it can or can’t infect a given species) (Davies & Pedersen, 2008, Pfenning-Butterworth et al., 2021) or intra-specific variability in infection-related traits (infectivity, resistance etc.) due to factors such as sex, genotype and environment (Vale et al., 2008, Lambrechts et al., 2006). This work thus advances on previous studies, while at the same time showing that sex differences in parasite load are not necessarily pervasive. References Behringer DC, Butler MJ, Shields JD (2006) Avoidance of disease by social lobsters. Nature, 441, 421–421. https://doi.org/10.1038/441421a Brierley L, Pedersen AB, Woolhouse MEJ (2019) Tissue tropism and transmission ecology predict virulence of human RNA viruses. PLOS Biology, 17, e3000206. https://doi.org/10.1371/journal.pbio.3000206 Davies TJ, Pedersen AB (2008) Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proceedings of the Royal Society B: Biological Sciences, 275, 1695–1701. https://doi.org/10.1098/rspb.2008.0284 Faria NR, Suchard MA, Rambaut A, Streicker DG, Lemey P (2013) Simultaneously reconstructing viral cross-species transmission history and identifying the underlying constraints. Philosophical Transactions of the Royal Society B: Biological Sciences, 368, 20120196. https://doi.org/10.1098/rstb.2012.0196 Gilbert GS, Webb CO (2007) Phylogenetic signal in plant pathogen–host range. Proceedings of the National Academy of Sciences, 104, 4979–4983. https://doi.org/10.1073/pnas.0607968104 Imrie RM, Roberts KE, Longdon B (2021) Between virus correlations in the outcome of infection across host species: Evidence of virus by host species interactions. Evolution Letters, 5, 472–483. https://doi.org/10.1002/evl3.247 Johnson PTJ, Rohr JR, Hoverman JT, Kellermanns E, Bowerman J, Lunde KB (2012) Living fast and dying of infection: host life history drives interspecific variation in infection and disease risk. Ecology Letters, 15, 235–242. https://doi.org/10.1111/j.1461-0248.2011.01730.x Kelly CD, Stoehr AM, Nunn C, Smyth KN, Prokop ZM (2018) Sexual dimorphism in immunity across animals: a meta-analysis. Ecology Letters, 21, 1885–1894. https://doi.org/10.1111/ele.13164 Lambrechts L, Chavatte J-M, Snounou G, Koella JC (2006) Environmental influence on the genetic basis of mosquito resistance to malaria parasites. Proceedings of the Royal Society B: Biological Sciences, 273, 1501–1506. https://doi.org/10.1098/rspb.2006.3483 Longdon B, Hadfield JD, Day JP, Smith SCL, McGonigle JE, Cogni R, Cao C, Jiggins FM (2015) The Causes and Consequences of Changes in Virulence following Pathogen Host Shifts. PLOS Pathogens, 11, e1004728. https://doi.org/10.1371/journal.ppat.1004728 Longdon B, Hadfield JD, Webster CL, Obbard DJ, Jiggins FM (2011) Host Phylogeny Determines Viral Persistence and Replication in Novel Hosts. PLOS Pathogens, 7, e1002260. https://doi.org/10.1371/journal.ppat.1002260 Pfenning-Butterworth AC, Davies TJ, Cressler CE (2021) Identifying co-phylogenetic hotspots for zoonotic disease. Philosophical Transactions of the Royal Society B: Biological Sciences, 376, 20200363. https://doi.org/10.1098/rstb.2020.0363 Roberts KE, Longdon B (2023) Heterogeneities in infection outcomes across species: examining sex and tissue differences in virus susceptibility. bioRxiv 2022.11.01.514663, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.11.01.514663 Roy BA, Kirchner JW (2000) Evolutionary Dynamics of Pathogen Resistance and Tolerance. Evolution, 54, 51–63. https://doi.org/10.1111/j.0014-3820.2000.tb00007.x Vale PF, Stjernman M, Little TJ (2008) Temperature-dependent costs of parasitism and maintenance of polymorphism under genotype-by-environment interactions. Journal of Evolutionary Biology, 21, 1418–1427. https://doi.org/10.1111/j.1420-9101.2008.01555.x | Heterogeneities in infection outcomes across species: sex and tissue differences in virus susceptibility | Katherine E Roberts, Ben Longdon | <p style="text-align: justify;">Species vary in their susceptibility to pathogens, and this can alter the ability of a pathogen to infect a novel host. However, many factors can generate heterogeneity in infection outcomes, obscuring our ability t... | | Evolutionary Ecology | Alison Duncan | Anonymous, Greg Hurst | 2022-11-03 11:17:42 | |
01 Mar 2021
Social Conflicts in Dictyostelium discoideum : A Matter of ScalesThe cell-level perspective in social conflicts in Dictyostelium discoideumRecommended by Jeremy Van Cleve based on reviews by Peter Conlin and ?The social amoeba Dictyostelium discoideum is an important model system for the study of cooperation and multicellularity as is has both unicellular and aggregative life phases. In the aggregative phase, which typically occurs when nutrients are limiting, individual cells eventually gather together to form a fruiting bodies whose spores may be dispersed to another, better, location and whose stalk cells, which support the spores, die. This extreme form of cooperation has been the focus of numerous studies that have revealed the importance genetic relatedness and kin selection (Hamilton 1964; Lehmann and Rousset 2014) in explaining the maintenance of this cooperative collective behavior (Strassmann et al. 2000; Kuzdzal-Fick et al. 2011; Strassmann and Queller 2011). However, much remains unknown with respect to how the interactions between individual cells, their neighbors, and their environment produce cooperative behavior at the scale of whole groups or collectives. In this preprint, Forget et al. (2021) describe how the D. discoideum system is crucial in this respect because it allows these cellular-level interactions to be studied in a systematic and tractable manner. References Forget, M., Adiba, S. and De Monte, S.(2021) Social conflicts in *Dictyostelium discoideum *: a matter of scales. HAL, hal-03088868, ver. 2 peer-reviewed and recommended by PCI Evolutionary Biology. https://hal.archives-ouvertes.fr/hal-03088868/ Hamilton, W. D. (1964). The genetical evolution of social behaviour. II. Journal of theoretical biology, 7(1), 17-52. doi: https://doi.org/10.1016/0022-5193(64)90039-6 Kuzdzal-Fick, J. J., Fox, S. A., Strassmann, J. E., and Queller, D. C. (2011). High relatedness is necessary and sufficient to maintain multicellularity in Dictyostelium. Science, 334(6062), 1548-1551. doi: https://doi.org/10.1126/science.1213272 Lehmann, L., and Rousset, F. (2014). The genetical theory of social behaviour. Philosophical Transactions of the Royal Society B: Biological Sciences, 369(1642), 20130357. doi: https://doi.org/10.1098/rstb.2013.0357 Strassmann, J. E., and Queller, D. C. (2011). Evolution of cooperation and control of cheating in a social microbe. Proceedings of the National Academy of Sciences, 108(Supplement 2), 10855-10862. doi: https://doi.org/10.1073/pnas.1102451108 Strassmann, J. E., Zhu, Y., & Queller, D. C. (2000). Altruism and social cheating in the social amoeba Dictyostelium discoideum. Nature, 408(6815), 965-967. doi: https://doi.org/10.1038/35050087 Thompson, C. R., & Kay, R. R. (2000). Cell-fate choice in Dictyostelium: intrinsic biases modulate sensitivity to DIF signaling. Developmental biology, 227(1), 56-64. doi: https://doi.org/10.1006/dbio.2000.9877 | Social Conflicts in Dictyostelium discoideum : A Matter of Scales | Forget, Mathieu; Adiba, Sandrine; De Monte, Silvia | <p>The 'social amoeba' Dictyostelium discoideum, where aggregation of genetically heterogeneous cells produces functional collective structures, epitomizes social conflicts associated with multicellular organization. 'Cheater' populations that hav... | | Behavior & Social Evolution, Evolutionary Dynamics, Evolutionary Theory, Experimental Evolution | Jeremy Van Cleve | 2020-08-28 10:37:21 | ||
29 Nov 2023
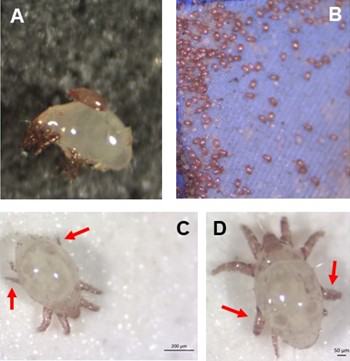
Individual differences in developmental trajectory leave a male polyphenic signature in bulb mite populationsWhat determines whether to scramble or fight in male bulb mitesRecommended by Ulrich Karl Steiner based on reviews by 2 anonymous reviewers
A classic textbook example in evolutionary ecology for phenotypic plasticity—the expression of different phenotypes by a genotype under different environmental conditions—is on Daphnia (Dodson 1989). If various species of this small crustacean are exposed to predation risk or cues thereof, their offspring show induced defense phenotypes including helmets, neck teeth, or head- and tail-spines. These induced morphological changes lower the risk of being eaten by a predator. As in Daphnia, induction can span over generations, while other induced phenotypic plastic changes are almost instantaneous, including many responses in physiology and behaviour (Gabriel et al. 2005). Larvae male bulb mites also show plasticity in morphologies throughout development. They can develop into a costly adult fighter morphology or a less costly, but vulnerable, scrambler type. The question Deere and Smallegange (Deere & Smallegange 2023) address is whether male bulb mite larvae can anticipate which type will likely be adaptive once they become adult, or alternatively, whether the resource availability or population density they experience during their larvae phase determines frequencies of adult scrambler and fighter types. They explore this question through experimental evolution, by removing different fractions of developing intermediate larva types. They thereby manipulate the stage structure of populations and alter selective forces on these stages. The potential shift of fixed genetics, imposed by the experimental selection regimes, is evaluated by fitness assays in green garden experiments. The exciting extension to classical experiments on phenotypic plasticity is that the authors aim at exploring eco-evolutionary feedbacks experimentally in a system that is a little more complex than basic host-parasite or predator-prey systems. The latter involve, for instance, rotifer-algae dynamics (Yoshida et al. 2003; Becks et al. 2012) or similar simple lab systems for which eco-evolutionary feedbacks have been demonstrated. The challenge for the exploration of more complex systems is revealed in the study by Deere and Smallegange. Their findings suggest that the frequencies of adult male morphotype is triggered by the environmental condition (nutrient availability) during the larval phase, i.e. a simple environmental induced plastic response. No fixed genetic shift in determining adult morphotype frequencies occurs. The trigger at the larva phase remains also not perfectly determined in their experiments, as population density and resource (food) availability are partly confounded. Additional complexity and selective aspects come into play in this system, as the targeted developmental stages that develop into fighter male morphs are also dispersal morphs. If selection on dispersal to avoid residing in food-limited environments is strong, triggering genetic shifts in fighter morphs by local population structure might be hard to experimentally achieve. Small sample sizes limit conclusions on complex interactions among duration of the experiment, population size, developmental stage types, and adult fighter frequencies. The presented study (Deere & Smallegange 2023) helps to bridge theoretical predictions and empirical evidence for eco-evolutionary feedbacks that goes beyond simple ecological-driven changes in population dynamics (Govaert et al. 2019).
References
Becks, L., Ellner, S.P., Jones, L.E. & Hairston, N.G. (2012). The functional genomics of an eco-evolutionary feedback loop: linking gene expression, trait evolution, and community dynamics. Ecol. Lett., 15, 492-501. | Individual differences in developmental trajectory leave a male polyphenic signature in bulb mite populations | Jacques A. Deere & Isabel M. Smallegange | <p style="text-align: justify;">Developmental plasticity alters phenotypes and can in that way change the response to selection. When alternative phenotypes show different life history trajectories, developmental plasticity can also affect, and be... | | Evolutionary Ecology, Life History, Phenotypic Plasticity, Sexual Selection | Ulrich Karl Steiner | 2023-02-07 12:14:33 | ||
22 Oct 2019

Geographic variation in adult and embryonic desiccation tolerance in a terrestrial-breeding frogTough as old boots: amphibians from drier habitats are more resistant to desiccation, but less flexible at exploiting wet conditionsRecommended by Ben Phillips based on reviews by Juan Diego Gaitan-Espitia, Jennifer Nicole Lohr and 1 anonymous reviewerSpecies everywhere are facing rapid climatic change, and we are increasingly asking whether populations will adapt, shift, or perish [1]. There is a growing realisation that, despite limited within-population genetic variation, many species exhibit substantial geographic variation in climate-relevant traits. This geographic variation might play an important role in facilitating adaptation to climate change [2,3]. References [1] Hoffmann, A. A., and Sgrò, C. M. (2011). Climate change and evolutionary adaptation. Nature, 470(7335), 479–485. doi: 10.1038/nature09670 | Geographic variation in adult and embryonic desiccation tolerance in a terrestrial-breeding frog | Rudin-Bitterli, T, Evans, J. P. and Mitchell, N. J. | <p>Intra-specific variation in the ability of individuals to tolerate environmental perturbations is often neglected when considering the impacts of climate change. Yet this information is potentially crucial for mitigating any deleterious effects... | | Adaptation, Evolutionary Applications, Evolutionary Ecology | Ben Phillips | 2018-05-07 03:35:08 | ||
09 Feb 2018
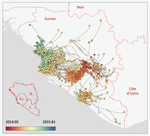
Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreakSimulating the effect of public health interventions using dated virus sequences and geographical dataRecommended by Samuel Alizon based on reviews by Christian Althaus, Chris Wymant and 1 anonymous reviewer
Perhaps because of its deadliness, the 2013-2016 Ebola Virus (EBOV) epidemics in West-Africa has led to unprecedented publication and sharing of full virus genome sequences. This was both rapid (90 full genomes were shared within weeks [1]) and important (more than 1500 full genomes have been released overall [2]). Furthermore, the availability of the metadata (especially GPS location) has led to depth analyses of the geographical spread of the epidemics [3]. References [1] Gire et al. 2014. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345: 1369–1372. doi: 10.1126/science.1259657. | Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreak | Simon Dellicour, Guy Baele, Gytis Dudas, Nuno R. Faria, Oliver G. Pybus, Marc A. Suchard, Andrew Rambaut, Philippe Lemey | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (https://doi.org/10.24072/pci.evolbiol.100046). The recent Ebola virus (EBOV) outbreak in West Africa witnessed considerable efforts to obtain viral genom... | | Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Samuel Alizon | 2017-09-30 13:49:57 |




