
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields▼ | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
16 Mar 2017

POSTPRINT
Correlated paternity measures mate monopolization and scales with the magnitude of sexual selectionMeasurement of sexual selection in plants made easierRecommended by Emmanuelle Porcher and Mathilde DufaySexual selection occurs in flowering plants too. However it tends to be understudied in comparison to animal sexual selection, in part because the minuscule size and long dispersal distances of the individuals producing male gametes (pollen grains) seriously complicate the estimation of male siring success and thereby the measurement of sexual selection. Dorken and Perry [1] introduce a novel and clever approach to estimate sexual selection in plants, which bypasses the need for a direct quantification of absolute male mating success. This approach builds on the fact that the strength of sexual selection is directly related to the ability of individuals to monopolize mates [2]. In plants, mate monopolization can be assessed by examining the proportion of seeds produced by a given plant that are full-sibs, i.e. that share the same father. A nice feature of this proportion of full-sib seeds per maternal parent is it equals the coefficient of correlated paternity of Ritland [3], which can be readily obtained from the hundreds of plant mating system studies using genetic markers. A less desirable feature of the proportion of full sibs per maternal plant is that it is inversely related to population size, an effect that should be corrected for. The resulting index of mate monopolization is a simple product: (coefficient of correlated paternity)x(population size – 1). The authors test whether their index of mate monopolization is a good correlate of sexual selection, measured more traditionally as the selection differential on a trait influencing mating success, using a combination of theoretical and experimental approaches. Both approaches confirm that the two quantities are positively correlated, which suggests that the index of mate monopolization could be a convenient way to estimate the relative strength of sexual selection in flowering plants. These results call for further investigation, e.g. to verify that the effect of population size is well controlled for, or to assess the effects of non-random mating and inbreeding depression; however, this work paves the way for an expansion of sexual selection studies in flowering plants. References [1] Dorken ME and Perry LE. 2017. Correlated paternity measures mate monopolization and scales with the magnitude of sexual selection. Journal of Evolutionary Biology 30: 377-387 doi: 10.1111/jeb.13013 [2] Klug H, Heuschele J, Jennions M and Kokko H. 2010. The mismeasurement of sexual selection. Journal of Evolutionary Biology 23:447-462. doi: 10.1111/j.1420-9101.2009.01921.x [3] Ritland K. 1989. Correlated matings in the partial selfer Mimulus guttatus. Evolution 43:848-859. doi: 10.2307/2409312 | Correlated paternity measures mate monopolization and scales with the magnitude of sexual selection | Dorken, ME and Perry LE | Indirect measures of sexual selection have been criticized because they can overestimate the magnitude of selection. In particular, they do not account for the degree to which mating opportunities can be monopolized by individuals of the sex that ... | | Sexual Selection | Emmanuelle Porcher | 2017-03-13 23:22:26 | ||
25 Jan 2024

Sperm production and allocation in response to risk of sperm competition in the black soldier fly Hermetia illucensElevated sperm production and faster transfer: plastic responses to the risk of sperm competition in males of the black sodier fly Hermetia illuceRecommended by Trine Bilde based on reviews by Rebecca Boulton, Isabel Smallegange and 1 anonymous reviewer based on reviews by Rebecca Boulton, Isabel Smallegange and 1 anonymous reviewer
In this paper (Manas et al., 2023), the authors investigate male responses to risk of sperm competition in the black soldier fly Hermetia illuce, a widespread insect that has gained recent attention for its potential to be farmed for sustainable food production (Tomberlin & van Huis, 2020). Using an experimental approach that simulated low-risk (males were kept individually) and high-risk (males were kept in groups of 10) of sperm competition, they found that males reared in groups showed a significant increase in sperm production compared with males reared individually. This shows a response to the rearing environment in sperm production that is consistent with an increase in the perceived risk of sperm competition. These males were then used in mating experiments to determine whether sperm allocation to females during mating was influenced by the perceived risk of sperm competition. Mating experiments were initiated in groups, since mating only occurs when more than one male and one female are present, indicating strong sexual selection in the wild. Once a copulation began, the pair was moved to a new environment with no competition, with male competitors, or with other females, to test how social environment and potentially the sex of surrounding individuals influenced sperm allocation during mating. Copulation duration and the number of sperm transferred were subsequently counted. In these mating experiments, the number of sperm stored in the female spermathecae increased under immediate risk of sperm competition. Interestingly, this was not because males copulated for longer depending on the risk of sperm competition, indicating that males respond plastically to the risk of competition by elevating their investment in sperm production and speed of sperm transfer. There was no difference between competitive environments consisting of males or females respectively, suggesting that it is the presence of other flies per se that influence sperm allocation. The study provides an interesting new example of how males alter reproductive investment in response to social context and sexual competition in their environment. In addition, it provides new insights into the reproductive biology of the black soldier fly Hermetia illucens, which may be relevant for optimizing farming conditions. References Manas F, Labrousse C, Bressac C (2023) Sperm production and allocation in response to risks of sperm competition in the black soldier fly Hermetia illucens. bioRxiv, 2023.06.20.544772, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.06.20.544772 Tomberlin JK, Van Huis A (2020) Black soldier fly from pest to ‘crown jewel’ of the insects as feed industry: an historical perspective. Journal of Insects as Food and Feed, 6, 1–4. https://doi.org/10.3920/JIFF2020.0003 | Sperm production and allocation in response to risk of sperm competition in the black soldier fly Hermetia illucens | Frédéric Manas, Carole Labrousse, Christophe Bressac | <p style="text-align: justify;">In polyandrous species, competition between males for offspring paternity goes on after copulation through the competition of their ejaculates for the fertilisation of female's oocytes. Given that males allocating m... | | Reproduction and Sex, Sexual Selection | Trine Bilde | 2023-06-26 09:41:07 | ||
11 Apr 2023
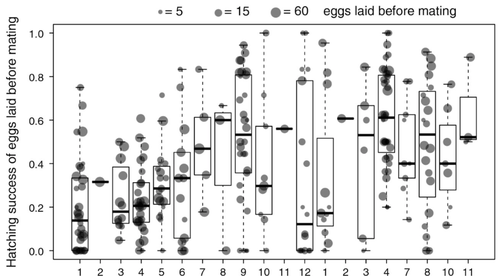
Facultative parthenogenesis: a transient state in transitions between sex and obligate asexuality in stick insects?Facultative parthenogenesis and transitions from sexual to asexual reproductionRecommended by Trine Bilde based on reviews by 3 anonymous reviewers
Despite a vast array of ways in which organisms can reproduce (Bell, 1982), most animals engage in sexual reproduction (Otto & Lenormand, 2002). A fascinating alternative to sex is parthenogenesis, where offspring are produced asexually from a gamete, typically the egg, without receiving genetic material from another gamete (Simon, Delmotte, Rispe, & Crease, 2003). One of the long-standing questions in the field is why parthenogenesis is not more widespread, given the costs associated with sex (Otto & Lenormand, 2002). Natural populations of most species appear to be reproducing either sexually or parthenogenetically, even if a species can employ both reproductive modes (Larose et al 2023). Larose et al (2023) highlight the conundrum in this pattern, as organisms that are capable of employing parthenogenesis facultatively would be able to gain the benefits of both modes of reproduction. Why then, is facultative parthenogenesis not more common? Larose et al (2023) propose that constraints on being efficient in both sexual and asexual reproduction could cause a trade-off between reproductive modes that favours an obligate strategy of either sex or no sex. This would provide an explanation for why facultative parthenogenesis is rare. Timema stick insects provide an excellent system to investigate reproductive strategies, as some species have parthenogenetic females, while other species are sexual, and they show repeated transitions from sexual reproduction to obligate parthenogenesis (Schwander & Crespi, 2009). The authors performed comprehensive and complementary studies in a recently discovered species T. douglasi, in which populations show both modes of reproduction, with some populations consisting only of females and others showing equal proportions of males and females. The sex ratio varied significantly, with the proportion of females ranging between 43-100% across 29 populations. These populations form a monophyletic clade with clustering into three genetic lineages and only a few cases of admixture. Females from all populations were capable of producing unfertilized eggs, but the hatching success varied hugely among populations and lineages (3-100%). Parthenogenetically produced offspring were homozygous, showing that parthenogenesis causes a complete loss of heterozygosity in a single generation. After producing eggs as virgins, females were mated to assess the capacity to also reproduce sexually, and fertilization increased the hatching success of eggs in two lineages. In one lineage, in which the hatching success of unfertilized eggs is similar to that of other sexually reproducing Timema species, fertilization reduced egg-hatching success, indicating a trade-off between reproductive modes with parthenogenetic reproduction performing best. Approximately 58% of the offspring produced after mating were fertilized, demonstrating the capacity of females to reproduce parthenogenetically also after mating has occurred, however with huge variation among individuals. This wonderful and meticulously performed study produces strong and complementary evidence for facultative parthenogenesis in T. douglasi populations. The study shows large variation in how reproductive mode is employed, supporting the existence of a trade-off between sexual and parthenogenetic reproduction. This might be an example of an ongoing transition from sexual to asexual reproduction, which indicates that obligate parthenogenesis may derive via transient facultative parthenogenesis. REFERENCES Bell, G. (1982) The Masterpiece of Nature: The Evolution and Genetics of Sexuality. University of California Press. 635 p. Otto, S. P., & Lenormand, T. (2002). Resolving the paradox of sex and recombination. Nature Reviews Genetics, 3(4), 252-261. https://doi.org/10.1038/nrg761 Schwander, T., & Crespi, B. J. (2009). Multiple direct transitions from sexual reproduction to apomictic parthenogenesis in Timema stick insects. Evolution, 63(1), 84-103. Simon, J.-C., Delmotte, F., Rispe, C., & Crease, T. (2003). Phylogenetic relationships between parthenogens and their sexual relatives: the possible routes to parthenogenesis in animals. Biological Journal of the Linnean Society, 79(1), 151-163. https://doi.org/10.1046/j.1095-8312.2003.00175.x Larose, C., Lavanchy, G., Freitas, S., Parker, D.J., Schwander, T. (2023) Facultative parthenogenesis: a transient state in transitions between sex and obligate asexuality in stick insects? bioRxiv, 2022.03.25.485836, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.03.25.485836 | Facultative parthenogenesis: a transient state in transitions between sex and obligate asexuality in stick insects? | Chloé Larose, Guillaume Lavanchy, Susana Freitas, Darren J. Parker, Tanja Schwander | <p>Transitions from obligate sex to obligate parthenogenesis have occurred repeatedly across the tree of life. Whether these transitions occur abruptly or via a transient phase of facultative parthenogenesis is rarely known. We discovered and char... | | Reproduction and Sex | Trine Bilde | 2022-05-20 10:41:13 | ||
25 Jan 2023
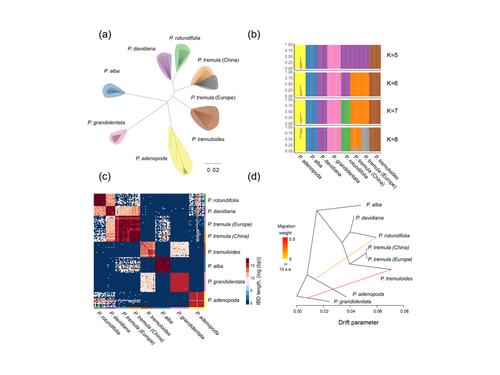
Drivers of genomic landscapes of differentiation across Populus divergence gradientShedding light on genomic divergence along the speciation continuumRecommended by Violaine Llaurens based on reviews by Camille Roux, Steven van Belleghem and 1 anonymous reviewer
The article “Drivers of genomic landscapes of differentiation across Populus divergence gradient” by Shang et al. describes an amazing dataset where genomic variations among 21 pairs of diverging poplar species are compared. Such comparisons are still quite rare and are needed to shed light on the processes shaping genomic divergence along the speciation gradient. Relying on two hundred whole-genome resequenced samples from 8 species that diverged from 1.3 to 4.8 million years ago, the authors aim at identifying the key factors involved in the genomic differentiation between species. They carried out a wide range of robust statistical tests aiming at characterizing the genomic differentiation along the genome of these species pairs. They highlight in particular the role of linked selection and gene flow in shaping the divergence along the genomes of species pairs. They also confirm the significance of introgression among species with a net divergence larger than the upper boundaries of the grey zone of speciation previously documented in animals (da from 0.005 to 0.02, Roux et al. 2016). Because these findings pave the way to research about the genomic mechanisms associated with speciation in species with allopatric and parapatric distributions, I warmingly recommend this article. References Roux C, Fraïsse C, Romiguier J, Anciaux Y, Galtier N, Bierne N (2016) Shedding Light on the Grey Zone of Speciation along a Continuum of Genomic Divergence. PLOS Biology, 14, e2000234. https://doi.org/10.1371/journal.pbio.2000234 Shang H, Rendón-Anaya M, Paun O, Field DL, Hess J, Vogl C, Liu J, Ingvarsson PK, Lexer C, Leroy T (2023) Drivers of genomic landscapes of differentiation across Populus divergence gradient. bioRxiv, 2021.08.26.457771, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.08.26.457771 | Drivers of genomic landscapes of differentiation across Populus divergence gradient | Huiying Shang, Martha Rendón-Anaya, Ovidiu Paun, View David L Field, Jaqueline Hess, Claus Vogl, Jianquan Liu, Pär K. Ingvarsson, Christian Lexer, Thibault Leroy | <p style="text-align: justify;">Speciation, the continuous process by which new species form, is often investigated by looking at the variation of nucleotide diversity and differentiation across the genome (hereafter genomic landscapes). A key cha... | | Population Genetics / Genomics, Speciation | Violaine Llaurens | 2021-09-06 14:12:27 | ||
04 Sep 2019

The discernible and hidden effects of clonality on the genotypic and genetic states of populations: improving our estimation of clonal ratesHow to estimate clonality from genetic data: use large samples and consider the biology of the speciesRecommended by Myriam Heuertz based on reviews by David Macaya-Sanz, Marcela Van Loo and 1 anonymous reviewer
Population geneticists frequently use the genetic and genotypic information of a population sample of individuals to make inferences on the reproductive system of a species. The detection of clones, i.e. individuals with the same genotype, can give information on whether there is clonal (vegetative) reproduction in the species. If clonality is detected, population geneticists typically use genotypic richness R, the number of distinct genotypes relative to the sample size, to estimate the rate of clonality c, which can be defined as the proportion of reproductive events that are clonal. Estimating the rate of clonality based on genotypic richness is however problematic because, to date, there is no analytical, nor simulation-based, characterization of this relationship. Furthermore, the effect of sampling on this relationship has never been critically examined. References [1] Stoeckel, S., Porro, B., and Arnaud-Haond, S. (2019). The discernible and hidden effects of clonality on the genotypic and genetic states of populations: improving our estimation of clonal rates. ArXiv:1902.09365 [q-Bio] v4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. Retrieved from http://arxiv.org/abs/1902.09365v4 | The discernible and hidden effects of clonality on the genotypic and genetic states of populations: improving our estimation of clonal rates | Solenn Stoeckel, Barbara Porro, Sophie Arnaud-Haond | <p>Partial clonality is widespread across the tree of life, but most population genetics models are conceived for exclusively clonal or sexual organisms. This gap hampers our understanding of the influence of clonality on evolutionary trajectories... | | Population Genetics / Genomics, Reproduction and Sex | Myriam Heuertz | 2019-02-28 10:10:56 | ||
14 Dec 2023

Genetic sex determination in three closely related hydrothermal vent gastropods, including one species with intersex individualsA shared XY sex chromosome system with variable recombination ratesRecommended by Tanja Schwander based on reviews by Hugo Darras, Daniel Jeffries and 1 anonymous reviewerMany species with separate sexes have evolved sex chromosomes, with the sex-limited chromosomes (i.e. the Y or W chromosomes) exhibiting a wide range of genetic divergences from their homologous X or Z chromosomes (Bachtrog et al., 2014). Variable divergences can result from the cessation of recombination between sex chromosomes that occurred at different time points, with the mechanisms of initiation and expansion of recombination suppression along sex chromosomes remaining poorly understood (Charlesworth, 2017). The study by Castel et al (2023) describes the serendipitous discovery of a shared XY sex chromosome system in three closely related hydrothermal vent gastropods. The X and Y chromosomes appear to still recombine but at variable rates across the three species. This variation makes the gastropod system a very promising focus for future research on sex chromosome evolution. An additional intriguing finding is that some females in one of three gastropod species contain male reproductive tissue in their gonads, providing a fascinating case of a mixed or transitory sexual system. Overall, the study by Castel et al (2023) offers the first insights into the reproduction and sex chromosome system of animals living in deep marine vents, which have remained poorly studied and open outstanding research perspectives on these creatures. References Bachtrog, D., J.E.Mank, C.L.Peichel, M.Kirkpatrick, S.P.Otto, T.L. Ashman, M.W.Hahn, J.Kitano, I.Mayrose, R.Ming, et al. 2014.Sex determination: why so many ways of doing it? PLoSBiol. 12:e1001899. https://doi.org/10.1371/journal.pbio.1001899 Charlesworth, D. Young sex chromosomes in plants and animals. 2019. New Phytologist 224: 1095–1107. https://doi.org/10.1111/nph.16002 Castel J, Pradillon F, Cueff V, Leger G, Daguin-Thiébaut C, Ruault S, Mary J, Hourdez S, Jollivet D, and Broquet T 2023. Genetic sex determination in three closely related hydrothermal vent gastropods, including one species with intersex individuals. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.04.11.536409 | Genetic sex determination in three closely related hydrothermal vent gastropods, including one species with intersex individuals | Castel J, Pradillon F, Cueff V, Leger G, Daguin-Thiébaut C, Ruault S, Mary J, Hourdez S, Jollivet D, and Broquet T | <p style="text-align: justify;">Molluscs have a wide variety of sexual systems and have undergone many transitions from separate sexes to hermaphroditism or vice versa, which is of interest for studying the evolution of sex determination and diffe... | | Population Genetics / Genomics, Reproduction and Sex | Tanja Schwander | 2023-04-14 11:48:25 | ||
12 Nov 2020
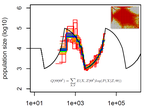
Limits and Convergence properties of the Sequentially Markovian CoalescentReview and Assessment of Performance of Genomic Inference Methods based on the Sequentially Markovian CoalescentRecommended by Stephan Schiffels based on reviews by 3 anonymous reviewers
The human genome not only encodes for biological functions and for what makes us human, it also encodes the population history of our ancestors. Changes in past population sizes, for example, affect the distribution of times to the most recent common ancestor (tMRCA) of genomic segments, which in turn can be inferred by sophisticated modelling along the genome. References [1] Li, H., and Durbin, R. (2011). Inference of human population history from individual whole-genome sequences. Nature, 475(7357), 493-496. doi: https://doi.org/10.1038/nature10231 | Limits and Convergence properties of the Sequentially Markovian Coalescent | Thibaut Sellinger, Diala Abu Awad, Aurélien Tellier | <p>Many methods based on the Sequentially Markovian Coalescent (SMC) have been and are being developed. These methods make use of genome sequence data to uncover population demographic history. More recently, new methods have extended the original... | | Population Genetics / Genomics | Stephan Schiffels | Anonymous | 2020-07-25 10:54:48 | |
09 Dec 2019

Systematics and geographical distribution of Galba species, a group of cryptic and worldwide freshwater snailsThe challenge of delineating species when they are hiddenRecommended by Fabien Condamine based on reviews by Pavel Matos, Christelle Fraïsse and Niklas WahlbergThe science of naming species (taxonomy) has been renewed with the developments of molecular sequencing, digitization of museum specimens, and novel analytical tools. However, naming species can be highly subjective, sometimes considered as an art [1], because it is based on human-based criteria that vary among taxonomists. Nonetheless, taxonomists often argue that species names are hypotheses, which are therefore testable and refutable as new evidence is provided. This challenge comes with a more and more recognized and critical need for rigorously delineated species not only for producing accurate species inventories, but more importantly many questions in evolutionary biology (e.g. speciation), ecology (e.g. ecosystem structure and functioning), conservation biology (e.g. targeting priorities) or biogeography (e.g. diversification processes) depend in part on those species inventories and our knowledge of species [2-3]. Inaccurate species boundaries or diversity estimates may lead us to deliver biased answers to those questions, exactly as phylogenetic trees must be reconstructed rigorously and analyzed critically because they are a first step toward discussing broader questions [2-3]. In this context, biological diversity needs to be studied from multiple and complementary perspectives requiring the collaboration of morphologists, molecular biologists, biogeographers, and modelers [4-5]. Integrative taxonomy has been proposed as a solution to tackle the challenge of delimiting species [2], especially in highly diverse and undocumented groups of organisms. References [1] Ohl, M. (2018). The art of naming. MIT Press. | Systematics and geographical distribution of Galba species, a group of cryptic and worldwide freshwater snails | Pilar Alda, Manon Lounnas, Antonio Alejandro Vázquez, Rolando Ayaqui, Manuel Calvopina, Maritza Celi-Erazo, Robert Dillon, Luisa Carolina González Ramírez, Eric S. Loker, Jenny Muzzio-Aroca, Alberto Orlando Nárvaez, Oscar Noya, Andrés Esteban Pere... | <p>Cryptic species can present a significant challenge to the application of systematic and biogeographic principles, especially if they are invasive or transmit parasites or pathogens. Detecting cryptic species requires a pluralistic approach in ... | | Phylogeography & Biogeography, Systematics / Taxonomy | Fabien Condamine | Pavel Matos, Christelle Fraïsse | 2019-05-25 10:34:57 | |
26 Oct 2021

Large-scale geographic survey provides insights into the colonization history of a major aphid pest on its cultivated apple host in Europe, North America and North AfricaThe evolutionary puzzle of the host-parasite-endosymbiont Russian doll for apples and aphidsRecommended by Ignacio Bravo based on reviews by Pedro Simões and 1 anonymous reviewerEach individual multicellular organism, each of our bodies, is a small universe. Every living surface -skin, cuticle, bark, mucosa- is the home place to milliards of bacteria, fungi and viruses. They constitute our microbiota. Some of them are essential for certain organisms. Other could not live without their hosts. For many species, the relationship between host and microbiota is so close that their histories are inseparable. The recognition of this biological inextricability has led to the notion of holobiont as the organism ensemble of host and microbiota. When individuals of a particular animal or plant species expand their geographical range, it is the holobiont that expands. And these processes of migration, expansion and colonization are often accompanied by evolutionary and ecological innovations in the interspecies relationships, at the macroscopic level (e.g. novel predator-prey or host-parasite interactions) and at the microscopic level (e.g. changes in the microbiota composition). From the human point of view, these novel interactions can be economically disastrous if they involve and threaten important crop or cattle species. And this is especially worrying in the present context of genetic standardization and intensification for mass-production on the one hand, and of climate change on the other. With this perspective, the international team led by Amandine Cornille presents a study aiming at understanding the evolutionary history of the rosy apple aphid Dysaphis plantaginea Passerini, a major pest of the cultivated apple tree Malus domestica Borkh (1). The apple tree was probably domesticated in Central Asia, and later disseminated by humans over the world in different waves, and it was probably introduced in Europe by the Greeks. It is however unclear when and where D. plantaginea started parasitizing the cultivated apple tree. The ancestral D. plantaginea could have already infected the wild ancestor of current cultivated apple trees, but the aphid is not common in Central Asia. Alternatively, it may have gained access only later to the plant, possibly via a host jump, from Pyrus to Malus that may have occurred in Asia Minor or in the Caucasus. In the present preprint, Olvera-Vázquez and coworkers have analysed over 650 D. plantaginea colonies from 52 orchards in 13 countries, in Western, Central and Eastern Europe as well as in Morocco and the USA. The authors have analysed the genetic diversity in the sampled aphids, and have characterized as well the composition of the associated endosymbiont bacteria. The analyses detect substantial recent admixture, but allow to identify aphid subpopulations slightly but significantly differentiated and isolated by distance, especially those in Morocco and the USA, as well as to determine the presence of significant gene flow. This process of colonization associated to gene flow is most likely indirectly driven by human interactions. Very interestingly, the data show that this genetic diversity in the aphids is not reflected by a corresponding diversity in the associated microbiota, largely dominated by a few Buchnera aphidicola variants. In order to determine polarity in the evolutionary history of the aphid-tree association, the authors have applied approximate Bayesian computing and machine learning approaches. Albeit promising, the results are not sufficiently robust to assess directionality nor to confidently assess the origin of the crop pest. Despite the large effort here communicated, the authors point to the lack of sufficient data (in terms of aphid isolates), especially originating from Central Asia. Such increased sampling will need to be implemented in the future in order to elucidate not only the origin and the demographic history of the interaction between the cultivated apple tree and the rosy apple aphid. This knowledge is needed to understand how this crop pest struggles with the different seasonal and geographical selection pressures while maintaining high genetic diversity, conspicuous gene flow, differentiated populations and low endosymbiontic diversity. References
| Large-scale geographic survey provides insights into the colonization history of a major aphid pest on its cultivated apple host in Europe, North America and North Africa | Olvera-Vazquez S.G., Remoué C., Venon A, Rousselet A., Grandcolas O., Azrine M., Momont L., Galan M., Benoit L., David G., Alhmedi A., Beliën T., Alins G., Franck P., Haddioui A., Jacobsen S.K., Andreev R., Simon S., Sigsgaard L., Guibert E., Tour... | <p style="text-align: justify;">With frequent host shifts involving the colonization of new hosts across large geographical ranges, crop pests are good models for examining the mechanisms of rapid colonization. The microbial partners of pest insec... | | Phylogeography & Biogeography, Population Genetics / Genomics, Species interactions | Ignacio Bravo | 2020-12-11 19:22:54 | ||
16 Mar 2023
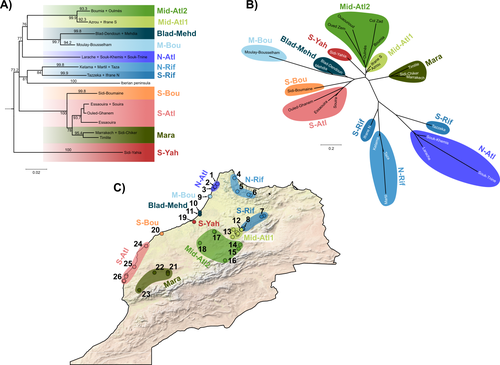
Phylogeographic breaks and how to find them: Separating vicariance from isolation by distance in a lizard with restricted dispersalThe difficult task of partitioning the effects of vicariance and isolation by distance in poor dispersersRecommended by Eric Pante based on reviews by Kevin Sánchez and Aglaia (Cilia) Antoniou
Partitioning the effects of vicariance and low dispersal has been a long-standing problem in historical biogeography and phylogeography. While the term “vicariance” refers to divergence in allopatry, caused by some physical (geological, geographical) or climatic barriers (e.g. Rosen 1978), isolation by distance refers to the genetic differentiation of remote populations due to the physical distance separating them, when the latter surpasses the scale of dispersal (Wright 1938, 1940, 1943). Vicariance and dispersal have long been considered as separate forces leading to separate scenarii of speciation (e.g. reviewed in Hickerson and Meyer 2008). Nevertheless, these two processes are strongly linked, as, for example, vicariance theory relies on the assumption that ancestral lineages were once linked by dispersal prior to physical or climatic isolation (Rosen 1978). Low dispersal and vicariance are not mutually exclusive, and distinguishing these two processes in heterogeneous landscapes, especially for poor dispersers, remains therefore a severe challenge. For example, low dispersal (and/or small population size) can give rise to geographic patterns consistent with a phylogeographic break and be mistaken for geographic isolation (Irwin 2002, Kuo and Avise 2005). The study of Rancilliac and colleagues (2023) is at the heart of this issue. It focuses on a nominal lizard species, the red-tailed spiny-footed lizard (Acanthodactylus erythrurus, Squamata: Lacertidae), which has a wide spatial distribution (from the Maghreb to the Iberian Peninsula), is found in a variety of different habitats, and has a wide range of morphological traits that do not always correlate with phylogeny. The main question is the following: have “the morphological and ecological diversification of this group been produced by vicariance and lineage diversification, or by local adaptation in the face of historical gene flow?” To tackle this question, the authors used sequence data from multiple mitochondrial and nuclear markers and a nested analysis workflow integrating phylogeography, multiple correspondence analyses and a relatively novel approach to IBD testing (Hausdorf & Henning, 2020). The latter is based on regression analysis and was shown to be less prone to error than the traditional (partial) Mantel test. While this set of methods allowed the partitioning of the effect of isolation by distance and vicariance in shaping contemporary genetic diversity in red-tailed spiny-footed lizards, some of the evolutionary history of this species complex remains blurred by ongoing gene flow and admixture, retention of ancestral polymorphism, or selection. The lack of congruence between mitochondrial and nuclear gene trees once again warns us that proposing evolutionary scenarii based on individual gene trees is a risky business. References Hausdorf B, Hennig C (2020) Species delimitation and geography. Molecular Ecology Resources, 20, 950–960. https://doi.org/10.1111/1755-0998.13184 Hickerson MJ, Meyer CP (2008) Testing comparative phylogeographic models of marine vicariance and dispersal using a hierarchical Bayesian approach. BMC Evolutionary Biology, 8, 322. https://doi.org/10.1186/1471-2148-8-322 Irwin DE (2002) Phylogeographic breaks without geographic barriers to gene flow. Evolution, 56, 2383–2394. https://doi.org/10.1111/j.0014-3820.2002.tb00164.x Kuo C-H, Avise JC (2005) Phylogeographic breaks in low-dispersal species: the emergence of concordance across gene trees. Genetica, 124, 179–186. https://doi.org/10.1007/s10709-005-2095-y Rancilhac L, Miralles A, Geniez P, Mendez-Aranda D, Beddek M, Brito JC, Leblois R, Crochet P-A (2023) Phylogeographic breaks and how to find them: An empirical attempt at separating vicariance from isolation by distance in a lizard with restricted dispersal. bioRxiv, 2022.09.30.510256, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.09.30.510256 Rosen DE (1978) Vicariant Patterns and Historical Explanation in Biogeography. Systematic Biology, 27, 159–188. https://doi.org/10.2307/2412970 Wright, S (1938) Size of population and breeding structure in relation to evolution. Science 87:430-431. Wright S (1940) Breeding Structure of Populations in Relation to Speciation. The American Naturalist, 74, 232–248. https://doi.org/10.1086/280891 Wright S (1943) Isolation by distance. Genetics, 28, 114–138. https://doi.org/10.1093/genetics/28.2.114 | Phylogeographic breaks and how to find them: Separating vicariance from isolation by distance in a lizard with restricted dispersal | Loïs Rancilhac, Aurélien Miralles, Philippe Geniez, Daniel Mendez-Arranda, Menad Beddek, José Carlos Brito, Raphaël Leblois, Pierre-André Crochet | <p>Aim</p> <p>Discontinuity in the distribution of genetic diversity (often based on mtDNA) is usually interpreted as evidence for phylogeographic breaks, underlying vicariant units. However, a misleading signal of phylogeographic break can arise... | | Phylogeography & Biogeography, Population Genetics / Genomics, Speciation, Systematics / Taxonomy | Eric Pante | Kevin Sánchez | 2022-10-05 13:11:28 |




