
BEDHOMME Stephanie
- Centre d'Ecologie Fonctionnelle et Evolutive, CNRS, Montpellier, France
- Adaptation, Experimental Evolution, Genome Evolution, Genotype-Phenotype, Life History
- recommender
Recommendations: 4
Reviews: 0
Recommendations: 4
A genomic amplification affecting a carboxylesterase gene cluster confers organophosphate resistance in the mosquito Aedes aegypti: from genomic characterization to high-throughput field detection
Identification of a gene cluster amplification associated with organophosphate insecticide resistance: from the diversity of the resistance allele complex to an efficient field detection assay
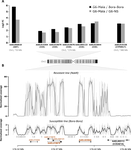
Recommended by Stephanie Bedhomme based on reviews by Diego Ayala and 2 anonymous reviewersThe emergence and spread of insecticide resistance compromises the efficiency of insecticides as prevention tool against the transmission of insect-transmitted diseases (Moyes et al. 2017). In this context, the understanding of the genetic mechanisms of resistance and the way resistant alleles spread in insect populations is necessary and important to envision resistance management policies. A common and important mechanism of insecticide resistance is gene amplification and in particular amplification of insecticide detoxification genes, which leads to the overexpression of these genes (Bass & Field, 2011). Cattel and coauthors (2020) adopt a combination of experimental approaches to study the role of gene amplification in resistance to organophosphate insecticides in the mosquito Aedes aegypti and its occurrence in populations of South East Asia and to develop a molecular test to track resistance alleles.
Their first approach consists in performing an artificial selection on laboratory Ae. Aegypti populations started with individuals collected in Laos. In the selected population, an initial 90% mortality by adult exposure to the organophosphate insecticide malathion is imposed. This population shows a steep increase in resistance to malathion and other organophosphate insecticides, which is absent in the paired control population. The transcriptomic patterns of the control and the evolved populations as well as of a reference sensitive population reveals, among other differences, the over-expression of five carboxy/choline esterase (CCE) genes in the insecticide selected population. These five genes happen to be clustered in the Ae. aegypti genome and whole genome sequencing of a highly resistant population combined to qPCR test on genomic DNA showed that the overexpression of these genes is due to gene amplification. Although it would have been more elegant to have replicate selected and control populations and to perform the transcriptomic and the genomic analyses directly on the experimental populations, the authors gather a set of experimental evidence which combined to previous knowledge on the function of the amplified and over-expressed genes and on their implication in organophosphate insecticide resistance in other species allow to discard the possibility that this gene amplification spread by drift in the selected population.
In a second part of the paper, copy number variation for CCE genes is checked in field sample populations. This test reveals the presence of resistance alleles in half of the fourteen South East Asia populations sampled. Very interestingly, it also reveals a high level of complexity and diversity among the resistance alleles: it shows first the existence, both in the experimental and the field populations, of at least two amplified alleles (differing by the number of genes amplified) and second a high variation in the copy number of amplified genes. This indicates that gene amplification as a molecular resistance mechanism has actually lead to a high diversity of resistance alleles. These alleles are likely to differ both by the level of resistance conferred and the fitness cost imposed in the absence of the insecticide and these two values are affecting the evolution of their frequency in the field and ultimately the spread of resistance.
The last part of the paper is devoted to the development of a high-throughput Taqman assay which allows to determine rapidly the copy number of one of the esterase genes amplified in the resistance alleles described earlier. This assay is nicely validated and will definitely be a useful tool to determine the occurrence of these resistance alleles in field population. The fact that it gives access to the copy number will also allow to follow its copy number across time and get insight into the complexity of resistance evolution by gene amplification.
To sum up, this paper studies the implication of carboxy/choline esterase genes amplification in organophosphate resistance evolution in Ae. aegypti, reveals the diversity among individuals and populations of this resistance mechanism, because of variation both in the identity of the genes amplified and in their copy number and sets up a fast and efficient tool to detect and follow the spread of these resistant alleles in the field. Additionally, the different experimental approaches adopted have generated genomic and transcriptomic data, of which only the part related to CCE gene amplification has been exploited. These data are very likely to reveal other genomic and expression determinants of resistance that will give access to an extra degree of complexity in organophosphate insecticide resistance determinism and evolution.
References
Bass C, Field LM (2011) Gene amplification and insecticide resistance. Pest Management Science, 67, 886–890. https://doi.org/10.1002/ps.2189
Cattel J, Haberkorn C, Laporte F, Gaude T, Cumer T, Renaud J, Sutherland IW, Hertz JC, Bonneville J-M, Arnaud V, Nous C, Fustec B, Boyer S, Marcombe S, David J-P (2020) A genomic amplification affecting a carboxylesterase gene cluster confers organophosphate resistance in the mosquito Aedes aegypti: from genomic characterization to high-throughput field detection. bioRxiv, 2020.06.08.139741, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. https://doi.org/10.1101/2020.06.08.139741
Moyes CL, Vontas J, Martins AJ, Ng LC, Koou SY, Dusfour I, Raghavendra K, Pinto J, Corbel V, David J-P, Weetman D (2017) Contemporary status of insecticide resistance in the major Aedes vectors of arboviruses infecting humans. PLOS Neglected Tropical Diseases, 11, e0005625. https://doi.org/10.1371/journal.pntd.0005625
Identifying drivers of parallel evolution: A regression model approach
A new statistical tool to identify the determinant of parallel evolution
Recommended by Stephanie Bedhomme based on reviews by Bastien Boussau and 1 anonymous reviewerIn experimental evolution followed by whole genome resequencing, parallel evolution, defined as the increase in frequency of identical changes in independent populations adapting to the same environment, is often considered as the product of similar selection pressures and the parallel changes are interpreted as adaptive.
However, theory predicts that heterogeneity both in mutation rate and selection intensity across the genome can trigger patterns of parallel evolution. It is thus important to evaluate and quantify the contribution of both mutation and selection in determining parallel evolution to interpret more accurately experimental evolution genomic data and also potentially improve our capacity to predict the genes that will respond to selection.
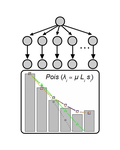
In their manuscript, Bailey, Guo and Bataillon [1] derive a framework of statistical models to partition the role of mutation and selection in determining patterns of parallel evolution at the gene level. The rationale is to use the synonymous mutations dataset as a baseline to characterize the mutation rate heterogeneity, assuming a negligible impact of selection on synonymous mutations and then analyse the non-synonymous dataset to identify additional source(s) of heterogeneity, by examining the proportion of the variation explained by a number of genomic variables.
This framework is applied to a published data set of resequencing of 40 Saccharomyces cerevisiae populations adapting to a laboratory environment [2]. The model explaining at best the synonymous mutations dataset is one of homogeneous mutation rate along the genome with a significant positive effect of gene length, likely reflecting variation in the size of the mutational target. For the non-synonymous mutations dataset, introducing heterogeneity between sites for the probability of a change to increase in frequency is improving the model fit and this heterogeneity can be partially explained by differences in gene length, recombination rate and number of functional protein domains.
The application of the framework to an experimental data set illustrates its capacity to disentangle the role of mutation and selection and to identify genomic variables explaining heterogeneity in parallel evolution probability but also points to potential limits, cautiously discussed by the authors: first, the number of mutations in the dataset analysed needs to be sufficient, in particular to establish the baseline on the synonymous dataset. Here, despite a high replication (40 populations evolved in the exact same conditions), the total number of synonymous mutations that could be analysed was not very high and there was only one case of a gene with synonymous mutation in two independent populations. Second, although the models are able to identify factors affecting the mutation counts, the proportion of the variation explained is quite low. The consequence is that the models correctly predicts the mutation count distribution but the objective of predicting on which genes the response to selection will occur still seems quite far away.
The framework developed in this manuscript [1] clearly represents a very useful tool for the analysis of large “evolve and resequence” data sets and to gain a better understanding of the determinants of parallel evolution in general. The extension of its application to mutations others than SNPs would provide the possibility to get a more complete picture of the differences in contributions of mutation and selection intensity heterogeneities depending on the mutation types.
References
[1] Bailey SF, Guo Q and Bataillon T (2018) Identifying drivers of parallel evolution: A regression model approach. bioRxiv 118695, ver. 4 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/118695
[2] Lang GI, Rice DP, Hickman, MJ, Sodergren E, Weinstock GM, Botstein D, and Desai MM (2013) Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature 500: 571–574. doi: 10.1038/nature12344
Things softly attained are long retained: Dissecting the Impacts of Selection Regimes on Polymorphism Maintenance in Experimental Spatially Heterogeneous Environments
Experimental test of the conditions of maintenance of polymorphism under hard and soft selection
Recommended by Stephanie Bedhomme based on reviews by Joachim Hermisson and 2 anonymous reviewers
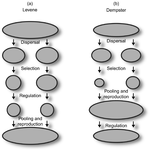
Theoretical work, initiated by Levene (1953) [1] and Dempster (1955) [2], suggests that within a given environment, the way populations are regulated and contribute to the next generation is a key factor for the maintenance of local adaptation polymorphism. In this theoretical context, hard selection describes the situation where the genetic composition of each population affects its contribution to the next generation whereas soft selection describes the case where the contribution of each population is fixed, whatever its genetic composition. Soft selection is able to maintain polymorphism, whereas hard selection invariably leads to the fixation of one of the alleles. Although the specific conditions (e.g. of migration between populations or drift level) in which this prediction holds have been studied in details by theoreticians, experimental tests have mainly failed, usually leading to the conclusion that the allele frequency dynamics was driven by other mechanisms in the experimental systems and conditions used. Gallet, Froissart and Ravigné [3] have set up a bacterial experimental system which allowed them to convincingly demonstrate that soft selection generates the conditions for polymorphism maintenance when hard selection does not, everything else being equal. The key ingredients of their experimental system are (1) the possibility to accurately produce hard and soft selection regimes when daily transferring the populations and (2) the ability to establish artificial well-characterized reproducible trade-offs. To do so, they used two genotypes resisting each one to one antibiotic and combined, across habitats, low antibiotic doses and difference in medium productivity. The experimental approach contains two complementary parts: the first one is looking at changes in the frequencies of two genotypes, initially introduced at around 50% each, over a small number of generations (ca 40) in different environments and selection regimes (soft/hard) and the second one is convincingly showing polymorphism protection by establishing that in soft selection regimes, the lowest fitness genotype is not eliminated even when introduced at low frequency. In this manuscript, a key point is the dialog between theoretical and experimental approaches. The experiments have been thought and designed to be as close as possible to the situations analysed in theoretical work. For example, the experimental polymorphism protection test (experiment 2) closely matches the equivalent analysis classically performed in theoretical approaches. This close fit between theory and experiment is clearly a strength of this study. This said, the experimental system allowing them to realise this close match also has some limitations. For example, changes in allele frequencies could only be monitored over a quite low number of generations because a longer time-scale would have allowed the contribution of de novo mutations and the likely emergence of a generalist genotype resisting to both antibiotics used to generate the local adaptation trade-offs. These limitations, as well as the actual significance of the experimental tests, are discussed in deep details in the manuscript.
References
[1] Levene H. 1953. Genetic equilibrium when more than one niche is available. American Naturalist 87: 331–333. doi: 10.1086/281792
[2] Dempster ER. 1955. Maintenance of genetic heterogeneity. Cold Spring Harbor Symposia on Quantitative Biology. 20: 25–32. doi: 10.1101/SQB.1955.020.01.005
[3] Gallet R, Froissart R, Ravigné V. 2017. Things softly attained are long retained: dissecting the impacts of selection regimes on polymorphism maintenance in experimental spatially heterogeneous environments. bioRxiv 100743; doi: 10.1101/100743

Experimental Evolution of Gene Expression and Plasticity in Alternative Selective Regimes
Genetic adaptation counters phenotypic plasticity in experimental evolution
Recommended by Luis-Miguel Chevin and Stephanie BedhommeHow do phenotypic plasticity and adaptive evolution interact in a novel or changing environment? Does evolution by natural selection generally reinforce initially plastic phenotypic responses, or does it instead oppose them? And to what extent does evolution of a trait involve evolution of its plasticity? These questions have lied at the heart of research on phenotypic evolution in heterogeneous environments ever since it was realized that the environment is likely to affect the expression of many (perhaps most) characters of an individual. Importantly, this broad definition of phenotypic plasticity as change in the average phenotype of a given genotype in response to its environment of development (or expression) does not involve any statement about the adaptiveness of the plastic changes. Theory on the evolution of plasticity has devoted much effort to understanding how reaction norm should evolve under different regimes of environmental change in space and time, and depending on genetic constraints on reaction norm shapes. However on an empirically ground, the questions above have mostly been addressed for individual traits, often chosen a priori for their likeliness to exhibit adaptive plasticity, and we still lack more systematic answers. These can be provided by so-called ‘phenomic’ approaches, where a large number of traits are tracked without prior information on their biological or ecological function. A problem is that the number of phenotypic characters that can be measured in an organism is virtually infinite (and to some extent arbitrary), and that scaling issues makes it difficult to compare different sets of traits. Gene-expression levels offer a partial solution to this dilemma, as they can be considered as a very large number of traits (one per typed gene) that can be measured easily and uniformly (fold change in the number of reads in RNAseq). As for any traits, expression levels of different genes may be genetically correlated, to an extent that depends on their regulation mechanism: cis-regulatory sequences that only affect expression of neighboring genes are likely to cause independent gene expression, while more systematic modifiers of expression (e.g. trans-regulators such as transcription factors) may cause correlated genetic responses of the expression of many genes. Huang and Agrawal [1] have studied plasticity and evolution of gene expression level in young larvae of populations of Drosophila melanogaster that have evolved for about 130 generations under either a constant environment (salt or cadmium), or an environment that is heterogeneous in time or space (combining salt and cadmium). They report a wealth of results, of which we summarize the most striking here. First, among genes that (i) were initially highly plastic and (ii) evolved significant divergence in expression levels between constant environment treatments, the evolved divergence is predominantly in the opposite direction to the initial plastic response. This suggests that either plasticity was initially maladaptive, or the selective pressure changed during the evolutionary process (see below). This somewhat unexpected result strikingly mirrors that from a study published last year in Nature [2], where the same pattern was found for responses of guppies to the presence of predators. However, Huang and Agrawal [1] went beyond this study by deciphering the underlying mechanisms in several interesting ways. First, they showed that change in gene expression often occurred at genes close to SNPs with differentiated frequencies across treatments (but not at genes with differentiated SNPs in their coding sequences), suggesting that cis-regulatory sequences are involved. This is also suggested by the fact that changes in gene expression are mostly caused by the increased expression of only one allele at polymorphic loci, and is a first step towards investigating the genetic underpinnings of (co)variation in gene expression levels. Another interesting set of findings concerns evolution of plasticity in treatments with variable environments. To compare the gene-expression plasticity that evolved in these treatments to an expectation, the authors considered that the expression levels in populations maintained for a long time under constant salt or cadmium had reached an optimum. The differences between these expression levels were thus assumed to predict the level of plasticity that should evolve in a heterogeneous environment (with both cadmium and salt) under perfect environmental predictability. The authors showed that plasticity did evolve more in the expected direction in heterogeneous than in constant environments, resulting in better adapted final expression levels across environments. Taken collectively, these results provide an unprecedented set of patterns that are greatly informative on how plasticity and evolution interact in constant versus changing environments. But of course, interpretations in terms of adaptive versus maladaptive plasticity are more challenging, as the authors themselves admit. Even though environmentally determined gene expression is the basic mechanism underlying the phenotypic plasticity of most traits, it is extremely difficult to relate to more integrated phenotypes for which we can understand the selection pressures, especially in multicellular organisms. The authors have recently investigated evolutionary change of quantitative traits in these selected lines, so it might be possible to establish links between reaction norms for macroscopic traits to those for gene expression levels. Such an approach would also involve tracking gene expression throughout life, rather than only in young larvae as done here, thus putting phenotypic complexity back in the picture also for expression levels. Another difficulty is that a plastic response that was originally adaptive may be replaced by an opposite evolutionary response in the long run, without having to invoke initially maladaptive plasticity. For instance, the authors mention the possibility that a generic stress response is initially triggered by cadmium, but is eventually unnecessary and costly after evolution of genetic mechanisms for cadmium detoxification (a case of so-called genetic accommodation). In any case, this study by Huang and Agrawal [1], together with the one by Ghalambor et al. last year [2], reports novel and unexpected results, which are likely to stimulate researchers interested in plasticity and evolution in heterogeneous environments for the years to come.
References
[1] Huang Y, Agrawal AF. 2016. Experimental Evolution of Gene Expression and Plasticity in Alternative Selective Regimes. PLoS Genetics 12:e1006336. doi: 10.1371/journal.pgen.1006336
[2] Ghalambor CK, Hoke KL, Ruell EW, Fischer EK, Reznick DN, Hughes KA. 2015. Non-adaptive plasticity potentiates rapid adaptive evolution of gene expression in nature. Nature 525: 372-375. doi: 10.1038/nature15256