
Recommendation
Perhaps because of its deadliness, the 2013-2016 Ebola Virus (EBOV) epidemics in West-Africa has led to unprecedented publication and sharing of full virus genome sequences. This was both rapid (90 full genomes were shared within weeks [1]) and important (more than 1500 full genomes have been released overall [2]). Furthermore, the availability of the metadata (especially GPS location) has led to depth analyses of the geographical spread of the epidemics [3].
In their work, Dellicour et al. [4] pursue earlier phylogeographical investigations in an original and yet simple approach to address questions of key public health importance. The originality of the approach is dual. First, from a technical standpoint, they capture the spread of infectious diseases in a continuous framework using a novel model that allows for rare long-distance dispersal events. Second, in a more classical discrete meta-population framework, they simulate the effect of public health interventions by pruning the phylogenetic tree and assessing how this affects key parameters. For instance, to simulate the effect of closing borders they remove subsets of the phylogeny that involved dispersal between countries and to simulate the effect of protecting a region by quarantine they remove all the leaves (i.e. the infections sampled) from this region. This phylogeny pruning is both original and simple. It is however limited because it currently assumes that policies are 100% effective and earlier modelling work on human influenza showed that long distance travel bans had to be implemented with >99% efficiency in order to slow epidemic growth from a time scale of days to weeks [5].
From a biological standpoint, Dellicour et al. [4] corroborate earlier findings that highly populated locations (>1,000,000 inhabitants) were crucial in explaining the magnitude of the epidemics but also show the importance of the transmission between the three capital cities. They also show that rare long-distance dispersing events of the virus are not key to explaining the magnitude of the epidemics (even though they assume 100% efficiency of suppressing long-distance event). Finally, thanks to their continuous model they estimate the speed of spread of the epidemics and are able to detect the effect of border closing on this speed.
Overall, this study [4], which involves state-of-the-art Bayesian inference methods of infection phylogenies using MCMC, stands out because of its effort to simulate public health interventions. It stands as an encouragement for the development of intervention models with increased realism and for even faster and larger virus sequence data sharing.
References
[1] Gire et al. 2014. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345: 1369–1372. doi: 10.1126/science.1259657.
[2] Holmes EC, Dudas G, Rambaut A and Andersen KG. 2016. The evolution of Ebola virus: insights from the 2013-2016 epidemic. Nature 538: 193–200. doi: 10.1038/nature19790.
[3] Dudas et al. 2017. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 544: 309–315 (2017). doi: 10.1038/nature22040.
[4] Dellicour S, Baele G, Dudas G, Faria NR, Pybus OG, Suchard MA, Rambaud A and Lemey P. 2018. Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreak. bioRxiv, 163691, ver. 3 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/163691.
[5] Hollingsworth TD, Ferguson NM and Anderson RM. 2006. Will travel restrictions control the international spread of pandemic influenza? Nature Medicine 12, 497–499. doi: 10.1038/nm0506-497.
DOI or URL of the preprint: 10.1101/163691
Version of the preprint: 2
Reply attached, thanks for the thorough evaluation!
 , posted 08 Feb 2018
, posted 08 Feb 2018The reviewers and I are very happy with this revised version.
I will be happy to write a recommendation.
In the meantime, one of the reviewers suggested some minor edits that could be worth adding.
I would like to thank the authors for revising their manuscript and adequately addressing my previous comments.
DOI or URL of the preprint: https://doi.org/10.1101/163691
Version of the preprint: 1
Dear Recommender, Please find uploaded our reply to the comments as well as a tracked changes version. The latter was produced using latex diff, which does not do a perfect job, but we hope it does reflect the major changes to the manuscript. Thank for handling our submission for recommendation and please do not hesitate to let us know if there are any remaining issues. Kind regards, Philippe
, posted 18 Jan 2018This work offer another very nice illustration of the power of recent advances in phylodynamics when applied to a dataset with dense sampling and rich meta-data (here the location of the infections). It focuses on the recent devastating ebola virus (EBOV) outbreak in West Africa and extends an earlier enormous analysis by Dudas et al. of the by adding a continuous phylogeography approach. It also refines the interpretation of the results by pinpointing the importance of the three capital cities in the magnitude of the outbreak.
Reviewer #1 made some very detailed suggestions and raised a general question about the interpretation of the tree prunning. He/she and Reviewer #2 also made suggestions to broaden the perspective of the article, for instance by discussing epidemiological studies that did not involve phylodynamics to estimate the spread of the epidemics.
In addition to the comments made by the reviewers, I have a couple of my own.
1) Would it be possible to provide confidence intervals for Figure 1 (there are some for panels E, F and G but only for the unprunned tree). The reason why I ask this is because it could help assess the magnitude of the effect. It could also explain why the curves in panels A and B increase at first (I was expecting a steady decrease).
2) Figure 1E is really beautiful! I was wondering if there is an explanation to the fact that recent case counts are below the inferred population size.
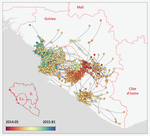
3) Figure 2 is also very nice but I expected to be able to find similarities because the sampled data should be the same. However, even the recent timepoints (in blue), which should all I guess be sampled, did not seem to be in the same place.
4) About the model choice (HKY+GAMMA and skygrid) the authors refer to Dudas et al. but it seems that the model choice is not really justified over there, e.g. testing for the most appropriate substitution model. If there is actual support, it would be worth mentionning it. Regarding the details about the priors, I guess the xml files will be made available?
The study by Dellicour and colleagues makes use of phylodynamic analyses for studying the spatial spread of Ebola during the 2013-2016 outbreak in West Africa. The authors extended their previously published phylogeographic framework to examine 1) the potential effect of intervention strategies - such as border closures - and 2) the process of spatial spread by introducing a continuous diffusion process (as opposed to the discrete approach in their earlier analysis). The methods are state-of-the-art and described in sufficient detail. The main findings of the study suggest that the Ebola epidemic was mainly driven by short- rather than long-distance dispersal. Furthermore, the study corroborates the notion that urban transmission was a major contributor to the characteristic spatial transmission dynamics that was observed in West Africa. I found the study rather technical and applying its findings in public health practice is maybe somewhat limited. However, the study is certainly a valuable contribution to the field of phylodynamics and provides an excellent example how genomic analyses can be used to infer the spatial spread of epidemics.
What I was missing a bit was a deeper discussion and comparison of the results to other studies outside the field of phylodynamics that investigated the spatial spread of Ebola and the impact of control interventions (e.g., border closures). The authors briefly mention two key papers using gravity-type models by Backer & Wallinga (ref. 27) and Kramer et al. (ref. 28). Others have estimated the velocity of Ebola spread at 1004 km per year (Zinszer et al., 2015, Lancet Infect Dis, PMID: 26333328) which seems to be in rough agreement with Fig. 3D. I also have a question related to how the authors call this velocity (mean dispersal distance per infection). Shouldn't it be per generation? In my view, it is not a single infection that spreads, but an epidemic that expands over subsequent generations.
Minor comments:
- Methods: The authors associate a random coordinate within the entire administrative area for sampled sequences that have the same geographic coordinates. I was wondering whether this assumption could introduce any sort of bias. For example, if all sequences came from exactly the same place in an otherwise large area, wouldn't associating random coordinates suggest wider spatial spread than what effectively happened?
- Fig. 1A: What is the dashed line on the peak of the distribution of lineage dispersal distances supposed to show?
- Fig. 1C: I could not find the dashed line that is described in the figure caption.
- Fig. 3: The word "sampled" appears twice in the second sentence.
- Fig. 4: I could not find a reference to this figure in the main text of the paper.
