
WYMANT Chris
- , ,
- Bioinformatics & Computational Biology, Evolutionary Epidemiology, Phylogenetics / Phylogenomics
Recommendations: 0
Reviews: 2
Reviews: 2

Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence data
Phylodynamics of hepatitis C virus reveals transmission dynamics within and between risk groups in Lyon
Recommended by David Rasmussen based on reviews by Chris Wymant and Louis DuPlessisGenomic epidemiology seeks to better understand the transmission dynamics of infectious pathogens using molecular sequence data. Phylodynamic methods have given genomic epidemiology new power to track the transmission dynamics of pathogens by combining phylogenetic analyses with epidemiological modeling. In recent year, applications of phylodynamics to chronic viral infections such as HIV and hepatitis C virus (HVC) have provided some of the best examples of how phylodynamic inference can provide valuable insights into transmission dynamics within and between different subpopulations or risk groups, allowing for more targeted interventions.
However, conducting phylodynamic inference under complex epidemiological models comes with many challenges. In some cases, it is not always straightforward or even possible to perform likelihood-based inference. Structured SIR-type models where infected individuals can belong to different subpopulations provide a classic example. In this case, the model is both nonlinear and has a high-dimensional state space due to tracking different types of hosts. Computing the likelihood of a phylogeny under such a model involves complex numerical integration or data augmentation methods [1]. In these situations, Approximate Bayesian Computation (ABC) provides an attractive alternative, as Bayesian inference can be performed without computing likelihoods as long as one can efficiently simulate data under the model to compare against empirical observations [2].
Previous work has shown how ABC approaches can be applied to fit epidemiological models to phylogenies [3,4]. Danesh et al. [5] further demonstrate the real world merits of ABC by fitting a structured SIR model to HCV data from Lyon, France. Using this model, they infer viral transmission dynamics between “classical” hosts (typically injected drug users) and “new” hosts (typically young MSM) and show that a recent increase in HCV incidence in Lyon is due to considerably higher transmission rates among “new” hosts . This study provides another great example of how phylodynamic analysis can help epidemiologists understand transmission patterns within and between different risk groups and the merits of expanding our toolkit of statistical methods for phylodynamic inference.
References
[1] Rasmussen, D. A., Volz, E. M., and Koelle, K. (2014). Phylodynamic inference for structured epidemiological models. PLoS Comput Biol, 10(4), e1003570. doi: https://doi.org/10.1371/journal.pcbi.1003570
[2] Beaumont, M. A., Zhang, W., and Balding, D. J. (2002). Approximate Bayesian computation in population genetics. Genetics, 162(4), 2025-2035.
[3] Ratmann, O., Donker, G., Meijer, A., Fraser, C., and Koelle, K. (2012). Phylodynamic inference and model assessment with approximate bayesian computation: influenza as a case study. PLoS Comput Biol, 8(12), e1002835. doi: https://doi.org/10.1371/journal.pcbi.1002835
[4] Saulnier, E., Gascuel, O., and Alizon, S. (2017). Inferring epidemiological parameters from phylogenies using regression-ABC: A comparative study. PLoS computational biology, 13(3), e1005416. doi: https://doi.org/10.1371/journal.pcbi.1005416
[5] Danesh, G., Virlogeux, V., Ramière, C., Charre, C., Cotte, L. and Alizon, S. (2020) Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence data. bioRxiv, 689158, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: https://doi.org/10.1101/689158
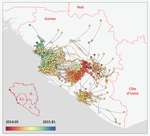
Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreak
Simulating the effect of public health interventions using dated virus sequences and geographical data
Recommended by Samuel Alizon based on reviews by Christian Althaus, Chris Wymant and 1 anonymous reviewerPerhaps because of its deadliness, the 2013-2016 Ebola Virus (EBOV) epidemics in West-Africa has led to unprecedented publication and sharing of full virus genome sequences. This was both rapid (90 full genomes were shared within weeks [1]) and important (more than 1500 full genomes have been released overall [2]). Furthermore, the availability of the metadata (especially GPS location) has led to depth analyses of the geographical spread of the epidemics [3].
In their work, Dellicour et al. [4] pursue earlier phylogeographical investigations in an original and yet simple approach to address questions of key public health importance. The originality of the approach is dual. First, from a technical standpoint, they capture the spread of infectious diseases in a continuous framework using a novel model that allows for rare long-distance dispersal events. Second, in a more classical discrete meta-population framework, they simulate the effect of public health interventions by pruning the phylogenetic tree and assessing how this affects key parameters. For instance, to simulate the effect of closing borders they remove subsets of the phylogeny that involved dispersal between countries and to simulate the effect of protecting a region by quarantine they remove all the leaves (i.e. the infections sampled) from this region. This phylogeny pruning is both original and simple. It is however limited because it currently assumes that policies are 100% effective and earlier modelling work on human influenza showed that long distance travel bans had to be implemented with >99% efficiency in order to slow epidemic growth from a time scale of days to weeks [5].
From a biological standpoint, Dellicour et al. [4] corroborate earlier findings that highly populated locations (>1,000,000 inhabitants) were crucial in explaining the magnitude of the epidemics but also show the importance of the transmission between the three capital cities. They also show that rare long-distance dispersing events of the virus are not key to explaining the magnitude of the epidemics (even though they assume 100% efficiency of suppressing long-distance event). Finally, thanks to their continuous model they estimate the speed of spread of the epidemics and are able to detect the effect of border closing on this speed.
Overall, this study [4], which involves state-of-the-art Bayesian inference methods of infection phylogenies using MCMC, stands out because of its effort to simulate public health interventions. It stands as an encouragement for the development of intervention models with increased realism and for even faster and larger virus sequence data sharing.
References
[1] Gire et al. 2014. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345: 1369–1372. doi: 10.1126/science.1259657.
[2] Holmes EC, Dudas G, Rambaut A and Andersen KG. 2016. The evolution of Ebola virus: insights from the 2013-2016 epidemic. Nature 538: 193–200. doi: 10.1038/nature19790.
[3] Dudas et al. 2017. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 544: 309–315 (2017). doi: 10.1038/nature22040.
[4] Dellicour S, Baele G, Dudas G, Faria NR, Pybus OG, Suchard MA, Rambaud A and Lemey P. 2018. Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreak. bioRxiv, 163691, ver. 3 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/163691.
[5] Hollingsworth TD, Ferguson NM and Anderson RM. 2006. Will travel restrictions control the international spread of pandemic influenza? Nature Medicine 12, 497–499. doi: 10.1038/nm0506-497.