
Recommendation
 based on reviews by 3 anonymous reviewers
based on reviews by 3 anonymous reviewers
Understanding the factors that shape the virome of a host is key to understanding virus ecology and evolution (Obbard, 2018; French & Holmes, 2020). There is still much to learn about the diversity and distribution of viruses in a host community (Wille et al., 2019; Chen et al., 2023). The viruses of parasitoid wasps are well studied, and their viruses, or integrated viral genes, are known to suppress their insect host’s immune response to enhance parasitoid survival (Herniou et al., 2013; Coffman et al., 2022). Likewise, the insect virome is being increasingly well studied (Shi et al., 2016), with the virome of Drosophila species being particularly well characterised over the best part of the last century (L'Heritier & Teissier, 1937; L'Heritier, 1970; Brun & Plus, 1980; Longdon et al., 2010; Longdon et al., 2011; Longdon et al., 2012; Webster et al., 2015; Webster et al., 2016; Medd et al., 2018; Wallace et al., 2021). However, the viromes of parasitoids and their insect host communities have been less well studied (Leigh et al., 2018; Caldas-Garcia et al., 2023), and the inherent connectivity between parasitoids and their hosts provides an interesting system to study virus host range and cross-species transmission.
Here, Varaldi et al (Varaldi et al., 2024) have examined the viruses associated with a community of nine Drosophilidae hosts and six parasitoids. Using both RNA and DNA sequencing of insects reared for two generations, they selected viruses that are maintained in the lab either via vertical transmission or contamination of rearing medium. From 55 pools of insects they found 53 virus-like sequences, 37 of which were novel. Parasitoids were host to nearly twice as many viruses as their Drosophila hosts, although they note this could be due to differences in the rearing temperatures of the hosts.
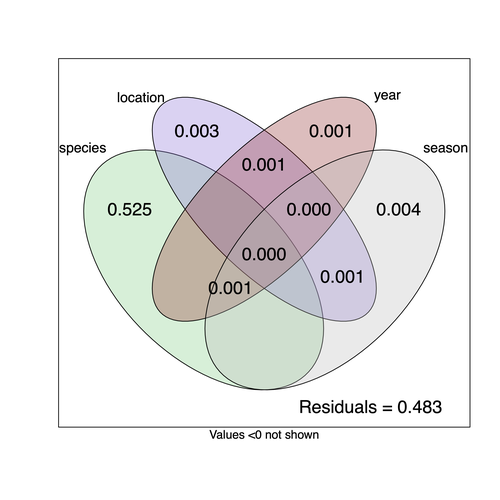
They next quantified if species, year, season, or location played a role in structuring the virome, finding only a significant effect of host species, which explained just over 50% of the variation in virus distribution. No evidence was found of related species sharing more similar virus communities. Although looking at a limited number of species, this suggests that these viruses are not co-speciating or preferentially host switching between closely related species.
Finally, they carried out crosses between lines of the parasitoid Leptopilina heterotoma that were infected and uninfected for a novel Iflavirus found in their sequencing data. They found evidence of high levels of maternal transmission and lower level horizontal transmission between wasp larvae parasitising the same host. No evidence of changes in parasitoid-induced mortality, developmental success or the sex ratio was found in iflavirus-infected parasitoids. Interestingly individuals infected with this RNA virus also contained viral DNA, but this did not appear to be integrated into the wasp genome.
Overall, this work has taken the first steps in examining the community structure of the virome of parasitoids together with their Drosophilidae hosts. This work will not doubt stimulate follow-up studies to explore the evolution and ecology of these novel virus communities.
References
Brun G, Plus N (1980) The viruses of Drosophila. In: The genetics and biology of Drosophila eds Ashburner M & Wright TRF), pp. 625-702. Academic Press, New York.
Caldas-Garcia GB, Santos VC, Fonseca PLC, de Almeida JPP, Costa MA, Aguiar ERGR (2023) The Viromes of Six Ecosystem Service Provider Parasitoid Wasps. Viruses, 15. https://doi.org/10.3390/v15122448
Chen YM, Hu SJ, Lin XD, Tian JH, Lv JX, Wang MR, Luo XQ, Pei YY, Hu RX, Song ZG, Holmes EC, Zhang YZ (2023) Host traits shape virome composition and virus transmission in wild small mammals. Cell, 186, 4662-4675 e4612. https://doi.org/10.1016/j.cell.2023.08.029
Coffman KA, Hankinson QM, Burke GR (2022) A viral mutualist employs posthatch transmission for vertical and horizontal spread among parasitoid wasps. Proceedings of the National Academy of Sciences of the United States of America, 119. https://doi.org/10.1073/pnas.2120048119
French RK, Holmes EC (2020) An Ecosystems Perspective on Virus Evolution and Emergence. Trends in Microbiology, 28, 165-175. https://doi.org/10.1016/j.tim.2019.10.010
Herniou EA, Huguet E, Thézé J, Bézier A, Periquet G, Drezen JM (2013) When parasitic wasps hijacked viruses: genomic and functional evolution of polydnaviruses. Philosophical Transactions of the Royal Society B-Biological Sciences, 368. https://doi.org/10.1098/rstb.2013.0051
L'Heritier PH (1970) Drosophila viruses and their role as evolutionary factors. Evolutionary Biology, 4, 185-209
L'Heritier PH, Teissier G (1937) Une anomalie physiologique héréditaire chez la Drosophile. C.R. Acad. Sci. Paris, 231, 192-194
Leigh BA, Bordenstein SR, Brooks AW, Mikaelyan A, Bordenstein SR (2018) Finer-Scale Phylosymbiosis: Insights from Insect Viromes. Msystems, 3. https://doi.org/10.1128/mSystems.00131-18
Longdon B, Obbard DJ, Jiggins FM (2010) Sigma viruses from three species of Drosophila form a major new clade in the rhabdovirus phylogeny. Proceedings of the Royal Society B, 277, 35-44.
https://doi.org/10.1098/rspb.2009.1472
Longdon B, Wilfert L, Jiggins FM (2012) The Sigma Viruses of Drosophila. Caister Academic Press, Norfolk, UK.
Longdon B, Wilfert L, Osei-Poku J, Cagney H, Obbard DJ, Jiggins FM (2011) Host switching by a vertically-transmitted rhabdovirus in Drosophila. Biology Letters, 7, 747-750.
https://doi.org/10.1098/rsbl.2011.0160
Medd NC, Fellous S, Waldron FM, Xuereb A, Nakai M, Cross JV, Obbard DJ (2018) The virome of Drosophila suzukii, an invasive pest of soft fruit. Virus Evol, 4, vey009.
https://doi.org/10.1093/ve/vey009
Obbard DJ (2018) Expansion of the metazoan virosphere: progress, pitfalls, and prospects. Curr Opin Virol, 31, 17-23. https://doi.org/10.1016/j.coviro.2018.08.008
Shi M, Lin XD, Tian JH, Chen LJ, Chen X, Li CX, Qin XC, Li J, Cao JP, Eden JS, Buchmann J, Wang W, Xu J, Holmes EC, Zhang YZ (2016) Redefining the invertebrate RNA virosphere. Nature. https://doi.org/10.1038/nature20167
Varaldi J, Lepetit D, Burlet N, Faber C, Baretje B, Allemand R (2024) Community structure of heritable viruses in a Drosophila-parasitoids complex. bioRxiv, 2023.2007.2029.551099, ver.3, peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.07.29.551099
Wallace MA, Coffman KA, Gilbert C, Ravindran S, Albery GF, Abbott J, Argyridou E, Bellosta P, Betancourt AJ, Colinet H, Eric K, Glaser-Schmitt A, Grath S, Jelic M, Kankare M, Kozeretska I, Loeschcke V, Montchamp-Moreau C, Ometto L, Onder BS, Orengo DJ, Parsch J, Pascual M, Patenkovic A, Puerma E, Ritchie MG, Rota-Stabelli O, Schou MF, Serga SV, Stamenkovic-Radak M, Tanaskovic M, Veselinovic MS, Vieira J, Vieira CP, Kapun M, Flatt T, Gonzalez J, Staubach F, Obbard DJ (2021) The discovery, distribution, and diversity of DNA viruses associated with Drosophila melanogaster in Europe. Virus Evol, 7, veab031. https://doi.org/10.1093/ve/veab031
Webster CL, Longdon B, Lewis SH, Obbard DJ (2016) Twenty-Five New Viruses Associated with the Drosophilidae (Diptera). Evol Bioinform Online, 12, 13-25. https://doi.org/10.4137/EBO.S39454
Webster CL, Waldron FM, Robertson S, Crowson D, Ferrai G, Quintana JF, Brouqui JM, Bayne EH, Longdon B, Buck AH, Lazzaro BP, Akorli J, Haddrill PR, Obbard DJ (2015) The discovery, distribution and evolution of viruses associated with Drosophila melanogaster. Plos Biology, 13(7): e1002210. https://doi.org/10.1371/journal.pbio.1002210
Wille M, Shi M, Klaassen M, Hurt AC, Holmes EC (2019) Virome heterogeneity and connectivity in waterfowl and shorebird communities. ISME J, 13, 2603-2616. https://doi.org/10.1038/s41396-019-0458-0
DOI or URL of the preprint: https://doi.org/10.1101/2023.07.29.551099
Version of the preprint: 2
Dear recommender,
Dear PCI reviewers,
We have done our best to address all the comments and minor issues raised by the three reviewers. We have included some additional analyses, such as testing for a host relatedness effect, as suggested by reviewer 3, and discussed the issue of intergenerational transmission of viruses in more detail. All other points have been carefully considered and we hope that this new version will be of sufficient quality to be accepted.
Below are the reviewers' comments, with our responses in bold.
In addition to the revised version deposited in BiorXiv (v3), we uploaded a pdf file highlighting the differences with the previous version available here :
https://filesender.renater.fr/?s=download&token=33cf8d8b-df6e-45f4-8fbd-a02f1467cd31.
All the best,
Julien
Reviews
Reviewed by anonymous reviewer, 05 Oct 2023 14:41
This manuscript describes structured sampling and metagenomic sequencing of Drosophila and their parasitoids in a framework that enables the testing of the factors that structure the whole virome. Because of the authors’ methods that include both DNA and RNA sequencing, and a multi-host species approach, this dataset is certainly unusual in the virus metagenomics literature. Additionally, I think this manuscript add significant value to the literature with its description of viruses in non-Dmel drosophilids and accompanying host range data. I found the manuscript generally well written, though some review of language needs doing (see below in point 1).
Some more general comments;
1) Introduction - I felt that the content was good and in particular you made good points on the lack on multi-species metagenomic virus discovery datasets. I don't feel that so much detail on heritable bacteria is needed here though, you could be more concise to get to the background on heritable viruses.
Since reviewer 3 was asking to develop this part to include a comparison with bacteria, on the contrary, we chose to develop slightly this part.
Additionally, there were several sentences in the introduction that need the English language reviewing, as there were a few linking words missing. For example; line 30 - I think this should to be changed to 'on the other hand', 'conversely', or something similar, rather than on the opposite. Also in line 31-33 - the English language in this sentence needs reviewing, some linking words are missing, and the sentence in line 41-43 needs reviewed.
We tried to reformulate.
2) I found the results section to be rather long, and much of this detail could probably be put into a supplementary document/table with details about each of the newly described viruses – however, I think this isn’t an essential change, it would just make the paper readable to a wider audience.
We agree that this part is still long despite our attempt to discuss them as quickly as possible by organizing their presentation by their main host drivers. However, we think placing all of this as supplementary would not be optimal, and chose to keep it in the main text. Nevertheless, we propose two ways of reading the manuscript to the reader, by adding this sentence at the beginning of section 3.5: “Readers who are primarily interested in general patterns, rather than the detailed composition of the viral community, may wish to skip to section 3.6.”
Some specific comments;
3) I think it would be good to more clearly state how exogenous and endogenous viral genomes were distinguished between in the contig assembly pipeline, as, as is acknowledged in the introduction both types might be present in this dataset.
The pipeline is designed to enrich for exogenous viral sequences in the sense that nucleic acids that would not be protected by a capsid are supposed to be eliminated (digested by nucleases). However, we cannot exclude that some insect DNA pass on anyway. If this happens, then we expect the presence of “eukaryotic-like” sequences to flank viral sequences. To test this, we blasted (blastn) each of the DNA contigs against nt with the expectation that if some of the contigs are in fact endogenized versions, then hits with insect genomes should be detected. None of the contigs revealed convincing evidence for eukaryotic sequence, with the exception of two of them. One is part of the twelve contigs assigned to Reoviridae1_D.sub_obs virus, with the first 88bp showing 98% identity with some Drosophila genomes (contig_9139). The other exception is one of the 11 contigs assigned to Vesantovirus_D.mel with 1020 first bp showing 99% identity with a retrotransposon gag protein from Drosophila (contig_22788). It is thus unclear whether these two contigs are endogenous or exogenous, but the overall picture is that the contigs reported here are not endogenized. This information is now given in the results section 3.2.
4) Could more details be given on the methods used to define a sample as infected by a particular viral contig in the analysis of factors structuring the viral community. Was there any limit used for the minimum number of reads mapping to a contig/or coverage depth for the sample to be considered infected? It seems to me that using coverage across 30% of the contig might still not be many reads? (section 3.3 results)
There was no filter on the number of reads for a sample to be considered as positive and indeed some samples had low reads number (minimum 2). However, the great majority of samples considered as positive did produce a relatively high number of reads (median 2399 and 166 respectively for DNA and RNA virus samples, and first quantile 670 and 35 respectively). More importantly, if we applied an additional minimal requirement of 10 reads per sample, the analysis produced very similar results with a strong species effect (0.527 instead of 0.525 and values <0.004 for the other components).
5) Fig. 2 – The x axis of this heatmap is almost impossible to read as its too small, could names be simplified and enlarged?
We increased the police to the maximum (+66%). We hope this is better now.
6) The formula, and packages and software used to glm mentioned in line 302, section 3.4 of the results should be given in the methods.
As this is a very basic analysis and the materials and methods section is already quite heavy, we decided to stick to the initial version.
7) The naming of viruses might need to be reconsidered for submission to genbank, as they might be considered not specific enough or in line with ICTV guidelines – after this submission the accession numbers should be added to this manuscript so that they can be easily associated with it.
The naming used in the manuscript is described in the header of each sequence we submitted to ncbi. However, the “organism” name we chose was slightly different to fit the ncbi nomenklatura (ex: the virus named Chuviridae1_D.im_n=3 in our manuscript was simply called “Chuviridae sp. 1” in the “organism” field and Chuviridae1_D.im_n=3 appears as a companion optional field).
8) In the discussion section, I would have liked to see more of a discussion of how the method of collecting and setting up 35 isofemale lines for each species, time and location might have influenced the viruses identified. Additionally, some more discussion of the host range observations for more than one virus would be interesting.
We added a sentence in the last part of the discussion indicating that our method of collecting probably enriched for viruses with low virulence.
The virus for which we discussed the host range issue was the one for which the clash was the most evident.
Reviewed by anonymous reviewer, 29 Sep 2023 08:36
This manuscript presents a very comprehensive analysis of heritable viruses circulating in a Drosophila-parasitoid wasps community. The authors combined an unbiased method to purify both DNA and RNA viruses with high-throughput sequencing and identified many viruses, a large proportion of them being new. While new viruses are being discovered at an accelerating rate in sequencing studies, the originality of the present work is the inclusion of multiple species known to interact in the wild. Characterizing viruses at this community level allowed the authors to more reliably define the host range for a given virus, compared to previous studies that may have focused on single species. In addition, their results provide interesting insights into the factors structuring viral diversity with the host species being by far the main driver and with parasitoid wasps carrying more viruses on average than their fly hosts. The authors conclude that this pattern is likely a consequence of the parasitoid lifestyle (while acknowledging that they cannot rule out a phylogenetic effect, since all parasitoids were Hymenoptera and all hosts were Diptera). The authors also report an intriguing observation: the presence of non-integrated DNA sequences matching the genome of an RNA virus in L. heterotoma (something that has previously been observed in other insects and interpreted as a mechanism of transgenerational antiviral immunity).
There is no doubt that this unique dataset will stimulate a number of follow-up studies aiming to characterize the ecological relevance and phenotypic impact of these new viruses, like the authors did for one of the viruses found in L. heterotoma.
My main comment relates to the definition of what a heritable virus is and perhaps this should be clarified somewhere. The authors seem to assume that all the viruses they found are heritable/vertically-transmitted, does that necessarily mean that they undergo transovarial or transmission through sperm? I agree that in principle bringing back insects to the lab and maintaining them for two generations should enrich for vertically-transmitted viruses but can we rule out the persistence of some non-heritable viruses during lab rearing? Most viruses transmitted through the environment will probably appear as heritable if we keep parents and offspring in a shared environment (even if generations do not overlap, depending on how long viral particles can survive outside their host). In this study, insect mothers from the field were used to establish isofemale lines in the lab and could have simply contaminated the media with virus particles, which in turn could infect the offspring emerging from the same vial (=shared environment). I guess what I’m trying to say here is that yes, those viruses were all transmitted from one generation to the next but this is in a very artificial and confined setting. How confident are we that, in the wild, all these viruses actually undergo frequent vertical transmission, in particular the ones infecting Drosophila? I’m not trying to diminish the work done here, I think it’s fantastic, but in my opinion future work will be needed to confirm which virus is “heritable” in an ecologically/evolutionary meaningful way.
We agree that we cannot rule out the possibility that some of the viruses we describe here are in fact pseudo-vertically transmitted in our experimental setting (not passed through gametes along generations but rather passed through a shared environment). And yes, future experiments should clarify this. Nevertheless, we can argue that at least part of their transmission may at be vertical in confined settings.
In natural settings, although we agree that the conditions are different, still, an egg-laying mother may contaminate the environment in which its offspring develop (decaying fruit). This surely does open lots of opportunities for horizontal transmission (because several unrelated egg-laying females may occupy the same patch of ressources) but some pseudo-vertical transmission may still contribute to viral transmission.
To clearly state this possibility, we added a few sentences in the introduction.
Other minor comments:
1) Line 13: I would suggest something like "the majority of which are not known to be pathogenic to their host" (since the phenotypic effects for most of them haven’t been investigated in details, they could still be very costly).
I agree with you and changed accordingly.
2) Line 44: “to accept laying eggs”
I corrected. Thank you.
3) Line 56-57: or interactions between these factors?
I added this notion.
4) Line 67-68: could there be an additional effect linked to the particular ecological relationship between Drosophila hosts and their parasitoids? Throughout evolution, viruses can jump to new host species when they come into contact. Wouldn’t we expect parasitoids to pick up more viruses from their Drosophila hosts if their interaction most frequently leads to the survival of the parasitoid and the death of the parasitized host? (whereas a host-shift in the opposite direction would be less likely)
I agree with this idea and have included it at the end of the introduction.
5) Line 82-84: perhaps this should be mentioned as a potential limitation of the study. Different viruses probably vary in their thermal tolerance, some might be outcompeted at certain temperatures while some may be favoured which could skew the difference in virus diversity between Drosophila hosts and parasitoids (since they were reared at different temperature). I’m also wondering what could be the effect of rearing the parasitoids on a laboratory line of D. melanogaster for two generations. For example, the fly immunity could have an effect on which viruses get passed on to the next parasitoid generation. Finally, do we know which viruses are present in the StFoy D. melanogaster line used for rearing all parasitoids? It doesn’t seem like this line was included in the sequencing experiment as a control (but I could have missed it). What is the negative control in figures 1 and 2?
Temperature: I agree that there is a possible confounding factor here. I added a few words on that possibility in the discussion section, but I also indicate that for the few viruses for which temperature effect on vertical transmission has been studied, there were either no temperature effect or an effect going in the opposite direction (less viral transmission at higher temperature).
We did not include the StFoy strain as a negative control. That would have been nice. However, we can argue that if this strain was infected by some viruses and the developing parasitoids were contaminated by some of them, then we would have expected the parasitoids (at least those belonging to the same species) to share the same viruses (from the same source). This was not the case.
The negative controls in Fig. 1 & 2 are water samples (I added this information in the legends).
6) Line 190: “Bioproject”.
Corrected thank you.
7) Line 196: “assemblies” instead of “assemblages”.
Corrected thank you.
8) Figure 1 & 2 legends: please indicate how the dendrograms were generated and what they represent (euclidian distances?)
I added this information.
9) Figure 3: maybe indicate in the legend that numbers are for percentages of total variance explained. The footnote indicates that values <0 are not shown. How can percentages of variance be negative? Or am I missing something?
The statistic used in the analysis (computed by the varpart function of the vegan package) is the adjusted R square. This statistic has been developed to remove the bias of raw R squares (the more explanatory variables or the more levels to a factor, the more spurious correlation is observed, Peres-Neto et al. 2006, https://doi.org/10.1890/0012-9658). The good properties of the corrected R square comes with the cost that sometimes, the adjusted R square becomes negative (which does not occur for the R2). This is likely happening when the true variance is very small. In that case, they were rounded to zero.
10) Line 371: remove empty parentheses?
I could not see the empty parentheses.
11) Line 477: shouldn’t it be D. kuntzei instead of D. phalerata according to figure 5 (phalerata is not in any of the heatmaps)? The shading of grey for lowly-infected species in figure 5 is rather difficult to follow in some cases, perhaps a different colour scheme would be preferable. It doesn't appear to me that the other species mentioned here (Trichopria, D. suboscura, etc...) are infected with LbTV by looking at the heatmap.
You are right! Thank you for identifying this mistake (D. kuntzei instead of D. phalerata).
The infection of Trichopria, D. suboscura, etc by LbTV is visible in Fig S5 where is indicated the “prevalence” of the virus in each species (based on the number of samples with coverage > 30%, independently of read numbers). However, the main driver of infection is likely to be L. boulardi if we rely on the relative number of reads produced per sample (as shown in Fig. 5).
12) Line 657-658: could some of these viruses not included in modules be coming from the D. melanogaster line used to maintain the different parasitoid species in the lab?
Again, we can argue that if this was the case, then we would expect that the different samples of the same parasitoid species share the same viruses (acquired from the same source). This was not the case (see in Fig. S3 for instance contig_6823 in some but not all L. heterotoma samples or contig_20830 in some but not all Trichopria samples).
13) Line 686: it looks like Phasmaviridae and Iflaviridae have been mistakenly inverted here when referring to maternal transmission.
Correct! Thank you for your vigilance!
Reviewed by anonymous reviewer, 04 Oct 2023 03:23
Summary:
The authors present a study in which wild Drosophila flies and Drosophila-attacking parasitoids were collected to determine the structure of viral communities associated with these insects from two different collection sites, years, and months. Heritable viruses were the focus of this study and were enriched by maintaining wild insects in the lab for two generations prior to sequencing. Four main types of sequencing libraries were generated from each of these samples: genomic DNA for COI amplicon sequencing and insect species confirmation, viral RNA random amplicon sequencing for RNA virus detection, viral DNA random amplicon sequencing for DNA virus detection, and a metagenomic library pooled from all samples to help establish a comprehensive virome database that viral reads from each sample could then be mapped onto. Protein sequence similarity was used to assign contigs to known viral taxa in order to obtain a consensus of virus species detected in each insect species, which included 53 viruses in total. The host species factor best explained the distribution of viruses detected across all samples, suggesting that an insect’s heritable virus community is distinct compared to those of other insect species, rather than influenced by location or time. In addition, the number of viral species detected in parasitoids was found to be higher than in Drosophila hosts, suggesting that parasitoids have a greater affinity for forming heritable virus associations. Lastly, functional experiments were performed to determine potential routes of transmission and phenotypes elicited by a new iflavirus found in L. heterotoma wasps.
Overall, I found the manuscript to be well-written, the methods were generally clear, and I think the results provide valuable new information regarding DNA and RNA viruses detected in this group of insects. My main comments speak to the lack of context presented for what virus community dynamics were expected from this dataset, and how additional background information on the study system and other microbial communities could have influenced these hypotheses.
Major Comments:
A missing link in this paper for me, which posits that viral communities have not been well-explored among interacting host species, is the relationships among the insects studied. Which parasitoid species attack which species of Drosophila in this community? Which parasitoid species are generalists vs specialists, what are the phylogenetic relationships among the various wasp and fly species collected, and how may these types of ecological and evolutionary relationships dictate what the authors expected to find regarding heritable viruses within each? I think a discussion of these dynamics would improve the manuscript, particularly in the Introduction and Discussion sections, as they are relevant to the goal of understanding what roles viruses play within interacting species.
We built a phylogeny for the species used (fig. 1) and also included a table summarizing the knowledge on host range of the five parasitoid species (table S1). We then tested for the effect of insect phylogeny on virome composition and found no effect. We did not include the test of the ecology (hosts or environment) because the host range of the wasps largely overlaps and is not fully known. Finally, as suggested, we refer now to literature on bacteria both in the introduction and discussion sections.
Related to the point above, I think the paper could benefit from a more in-depth dialogue of what factors the authors hypothesized might shape the viral communities in these insects and why. It is briefly mentioned in the Discussion section that the results support the evolution of specificity for these heritable viruses that leads to their infection of a single host, but I think the authors’ a priori expectations should also be discussed in the Introduction to contextualize the goals of the study. For example, the authors mention the wealth of information we now have on the ways heritable bacteria impact insect biology, while we know relatively little on how viruses shape these same communities. I think pertinent information in the Introduction could therefore be to indicate what factors we know impact heritable bacterial communities in insects, which could be the basis for hypotheses of whether viruses follow similar trends.
We added a few sentences to present the main factors shaping bacteria communities in insects (host phylogeny). We tested the effect of insect phylogeny on viral communities (3.4) and discussed the results in the light of the knowledge acquired on bacterial communities.
In general, I think the Introduction could be more succinct in several places, especially if adding more context to the study system and the hypotheses that prompted this work. For example, the second and third paragraphs (starting on L9 and L23) could be condensed into a single paragraph, since they share a similar theme (i.e. we know little of heritable virus communities and their interactions). Similarly, introductory paragraphs 4 and 5 (starting on L38 and L58) could be condensed into one paragraph that summarizes briefly the work presented in this study.
The second and the third paragraph do not exactly address the same issue. The 2d one focuses on the extant of the unknown viral diversity, whereas the third addresses the question of the structuration of this diversity. Similarly, the 4th and 5th do address different ideas: the 4th present the structuration issue, while the 5th presents the idea that parasitoids are expected to have more viruses than their hosts.
Is it possible that insect species was the greatest structuring factor to virus communities, due in part to how many different species were included (n = 15) in the analysis and how few variations comprised the other factors tested (n = 2) (e.g. only two different locations, two different months, and two different years)? If more locations and/or more time points were sampled, would this increase the authors’ ability to accurately determine which factor(s) explain the distribution of viruses identified? If so, I think this should be addressed in the Discussion.
The statistic used in the analysis (computed by the varpart function of the vegan package) is the adjusted R square. This statistic has been precisely developed to remove the bias you pointed at (the more explanatory variables or the more levels to a factor, the more spurious correlation is observed). This bias and the good properties of the corrected R square are given in the following reference (see fig. 2):
Peres-Neto, Pedro R., Pierre Legendre, Stéphane Dray, & Daniel Borcard. « Variation Partitioning of Species Data Matrices: Estimation and Comparison of Fractions ». Ecology 87, no 10 (2006): 2614‑25. https://doi.org/10.1890/0012-9658(2006)87[2614:vposdm]2.0.co;2.
We refer to this issue (and its resolution) in the figure legend.
Minor Comments:
L76: Please elaborate here on what the climate differences exist between the two locations.
We already indicate that the climates are Mediterranean in Gotheron and Continental in Ige. We do think that this minimal information is sufficient, especially because no effect has been ascribable to geographic location.
L81: Can the authors include more information on how these isofemale lines were maintained for two generations in the lab? Specifically, is it possible that horizontal transmission of these viruses could have occurred among individuals of the same line, or between parasitoids and D. melanogaster host flies during these two generations? Is the lab strain of D. melanogaster free of viruses or could these host flies be contributing to the viral communities that ultimately were sequenced from wasps?
Isofemale lines were maintained in small groups, but adults were removed after egg laying. So, pseudo vertical transmission through contamination of the medium is a possibility.
As reported above, the D. mel lab strain was not tested for viruses. However, we can argue that if the viruses we show here derived from viruses infecting this D.mel strain, then we would expect the different samples of the same parasitoid species to share the same viruses (acquired from the same source). This was not the case (see in Fig. S3 for instance contig_6823 in some but not all L. heterotoma samples or contig_20830 in some but not all Trichopria samples).
L232: How was superparasitism between uninfected female wasps and infected male wasps ensured? If some proportion of fly larvae were initially infested with an uninfected female wasp egg but received no supernumerary wasp eggs laid by virus-infected wasp mothers, the resulting female wasp offspring would still be uninfected, due to no exposure to the virus during parasitism. This scenario, if not checked, could lead to an underestimation of horizontal virus transmission rates, which I think should be reported in the results.
I agree with that and added a sentence in the result section.
Figures 1 and 2: I found these heatmaps quite difficult to understand initially, due to the minimal description of their contents in the figure legends and the manuscript text. I think more detailed information should be added to each figure legend, such as what the different clustering axes refer to. I also think a summary of the results conveyed by these maps should be included in the Results section, such as after their first mention on L279.
We tried to clarify the legend.
L686: Phasmaviridae is used in this sentence, although I believe the experiments were performed on the iflavirus, correct?
Correct. Thank you for your vigilance.
, posted 09 Oct 2023, validated 09 Oct 2023Dear Julien et al,
Sorry this has taken so long - in the end you've ended up with three constructive reviews from experts in the field who are very well placed to comment on the work. All three of the reviewers appreciated the neat approach taken to examine the community structure of the virome of Drosophila and their parasitoids. The reviewers (and myself) were in agreement that this is a useful and interesting set of results, and a nicely wirtten manuscript, and will not doubt stimulate follow up studies to explore the evolution and ecology of these novel virus communities. There are a range of constructive comments that I hope are minimal effort to address, and I think will improve the manuscript. In particular I concur with reviewer 2's comments regarding whether these viruses are truly vertically transmitted (under the definition of transmitted through eggs or sperm) or passed to offspring horizontally (eg through contaminated food)? Any queries let me know.
All the best,
Ben Longdon
This manuscript describes structured sampling and metagenomic sequencing of Drosophila and their parasitoids in a framework that enables the testing of the factors that structure the whole virome. Because of the authors’ methods that include both DNA and RNA sequencing, and a multi-host species approach, this dataset is certainly unusual in the virus metagenomics literature. Additionally, I think this manuscript add significant value to the literature with its description of viruses in non-Dmel drosophilids and accompanying host range data. I found the manuscript generally well written, though some review of language needs doing (see below in point 1).
Some more general comments;
1) Introduction - I felt that the content was good and in particular you made good points on the lack on multi-species metagenomic virus discovery datasets. I don't feel that so much detail on heritable bacteria is needed here though, you could be more concise to get to the background on heritable viruses. Additionally, there were several sentences in the introduction that need the English language reviewing, as there were a few linking words missing. For example; line 30 - I think this should to be changed to 'on the other hand', 'conversely', or something similar, rather than on the opposite. Also in line 31-33 - the English language in this sentence needs reviewing, some linking words are missing, and the sentence in line 41-43 needs reviewed.
2) I found the results section to be rather long, and much of this detail could probably be put into a supplementary document/table with details about each of the newly described viruses – however, I think this isn’t an essential change, it would just make the paper readable to a wider audience.
Some specific comments;
3) I think it would be good to more clearly state how exogenous and endogenous viral genomes were distinguished between in the contig assembly pipeline, as, as is acknowledged in the introduction both types might be present in this dataset.
4) Could more details be given on the methods used to define a sample as infected by a particular viral contig in the analysis of factors structuring the viral community. Was there any limit used for the minimum number of reads mapping to a contig/or coverage depth for the sample to be considered infected? It seems to me that using coverage across 30% of the contig might still not be many reads? (section 3.3 results)
5) Fig. 2 – The x axis of this heatmap is almost impossible to read as its too small, could names be simplified and enlarged?
6) The formula, and packages and software used to glm mentioned in line 302, section 3.4 of the results should be given in the methods.
7) The naming of viruses might need to be reconsidered for submission to genbank, as they might be considered not specific enough or in line with ICTV guidelines – after this submission the accession numbers should be added to this manuscript so that they can be easily associated with it.
8) In the discussion section, I would have liked to see more of a discussion of how the method of collecting and setting up 35 isofemale lines for each species, time and location might have influenced the viruses identified. Additionally, some more discussion of the host range observations for more than one virus would be interesting.
https://doi.org/10.24072/pci.evolbiol.100701.rev11This manuscript presents a very comprehensive analysis of heritable viruses circulating in a Drosophila-parasitoid wasps community. The authors combined an unbiased method to purify both DNA and RNA viruses with high-throughput sequencing and identified many viruses, a large proportion of them being new. While new viruses are being discovered at an accelerating rate in sequencing studies, the originality of the present work is the inclusion of multiple species known to interact in the wild. Characterizing viruses at this community level allowed the authors to more reliably define the host range for a given virus, compared to previous studies that may have focused on single species. In addition, their results provide interesting insights into the factors structuring viral diversity with the host species being by far the main driver and with parasitoid wasps carrying more viruses on average than their fly hosts. The authors conclude that this pattern is likely a consequence of the parasitoid lifestyle (while acknowledging that they cannot rule out a phylogenetic effect, since all parasitoids were Hymenoptera and all hosts were Diptera). The authors also report an intriguing observation: the presence of non-integrated DNA sequences matching the genome of an RNA virus in L. heterotoma (something that has previously been observed in other insects and interpreted as a mechanism of transgenerational antiviral immunity).
There is no doubt that this unique dataset will stimulate a number of follow-up studies aiming to characterize the ecological relevance and phenotypic impact of these new viruses, like the authors did for one of the viruses found in L. heterotoma.
My main comment relates to the definition of what a heritable virus is and perhaps this should be clarified somewhere. The authors seem to assume that all the viruses they found are heritable/vertically-transmitted, does that necessarily mean that they undergo transovarial or transmission through sperm? I agree that in principle bringing back insects to the lab and maintaining them for two generations should enrich for vertically-transmitted viruses but can we rule out the persistence of some non-heritable viruses during lab rearing? Most viruses transmitted through the environment will probably appear as heritable if we keep parents and offspring in a shared environment (even if generations do not overlap, depending on how long viral particles can survive outside their host). In this study, insect mothers from the field were used to establish isofemale lines in the lab and could have simply contaminated the media with virus particles, which in turn could infect the offspring emerging from the same vial (=shared environment). I guess what I’m trying to say here is that yes, those viruses were all transmitted from one generation to the next but this is in a very artificial and confined setting. How confident are we that, in the wild, all these viruses actually undergo frequent vertical transmission, in particular the ones infecting Drosophila? I’m not trying to diminish the work done here, I think it’s fantastic, but in my opinion future work will be needed to confirm which virus is “heritable” in an ecologically/evolutionary meaningful way.
Other minor comments:
1) Line 13: I would suggest something like "the majority of which are not known to be pathogenic to their host" (since the phenotypic effects for most of them haven’t been investigated in details, they could still be very costly).
2) Line 44: “to accept laying eggs”
3) Line 56-57: or interactions between these factors?
4) Line 67-68: could there be an additional effect linked to the particular ecological relationship between Drosophila hosts and their parasitoids? Throughout evolution, viruses can jump to new host species when they come into contact. Wouldn’t we expect parasitoids to pick up more viruses from their Drosophila hosts if their interaction most frequently leads to the survival of the parasitoid and the death of the parasitized host? (whereas a host-shift in the opposite direction would be less likely)
5) Line 82-84: perhaps this should be mentioned as a potential limitation of the study. Different viruses probably vary in their thermal tolerance, some might be outcompeted at certain temperatures while some may be favoured which could skew the difference in virus diversity between Drosophila hosts and parasitoids (since they were reared at different temperature). I’m also wondering what could be the effect of rearing the parasitoids on a laboratory line of D. melanogaster for two generations. For example, the fly immunity could have an effect on which viruses get passed on to the next parasitoid generation. Finally, do we know which viruses are present in the StFoy D. melanogaster line used for rearing all parasitoids? It doesn’t seem like this line was included in the sequencing experiment as a control (but I could have missed it). What is the negative control in figures 1 and 2?
6) Line 190: “Bioproject”.
7) Line 196: “assemblies” instead of “assemblages”.
8) Figure 1 & 2 legends: please indicate how the dendrograms were generated and what they represent (euclidian distances?)
9) Figure 3: maybe indicate in the legend that numbers are for percentages of total variance explained. The footnote indicates that values <0 are not shown. How can percentages of variance be negative? Or am I missing something?
10) Line 371: remove empty parentheses?
11) Line 477: shouldn’t it be D. kuntzei instead of D. phalerata according to figure 5 (phalerata is not in any of the heatmaps)? The shading of grey for lowly-infected species in figure 5 is rather difficult to follow in some cases, perhaps a different colour scheme would be preferable. It doesn't appear to me that the other species mentioned here (Trichopria, D. suboscura, etc...) are infected with LbTV by looking at the heatmap.
12) Line 657-658: could some of these viruses not included in modules be coming from the D. melanogaster line used to maintain the different parasitoid species in the lab?
13) Line 686: it looks like Phasmaviridae and Iflaviridae have been mistakenly inverted here when referring to maternal transmission.
https://doi.org/10.24072/pci.evolbiol.100701.rev12Summary:
The authors present a study in which wild Drosophila flies and Drosophila-attacking parasitoids were collected to determine the structure of viral communities associated with these insects from two different collection sites, years, and months. Heritable viruses were the focus of this study and were enriched by maintaining wild insects in the lab for two generations prior to sequencing. Four main types of sequencing libraries were generated from each of these samples: genomic DNA for COI amplicon sequencing and insect species confirmation, viral RNA random amplicon sequencing for RNA virus detection, viral DNA random amplicon sequencing for DNA virus detection, and a metagenomic library pooled from all samples to help establish a comprehensive virome database that viral reads from each sample could then be mapped onto. Protein sequence similarity was used to assign contigs to known viral taxa in order to obtain a consensus of virus species detected in each insect species, which included 53 viruses in total. The host species factor best explained the distribution of viruses detected across all samples, suggesting that an insect’s heritable virus community is distinct compared to those of other insect species, rather than influenced by location or time. In addition, the number of viral species detected in parasitoids was found to be higher than in Drosophila hosts, suggesting that parasitoids have a greater affinity for forming heritable virus associations. Lastly, functional experiments were performed to determine potential routes of transmission and phenotypes elicited by a new iflavirus found in L. heterotoma wasps.
Overall, I found the manuscript to be well-written, the methods were generally clear, and I think the results provide valuable new information regarding DNA and RNA viruses detected in this group of insects. My main comments speak to the lack of context presented for what virus community dynamics were expected from this dataset, and how additional background information on the study system and other microbial communities could have influenced these hypotheses.
Major Comments:
A missing link in this paper for me, which posits that viral communities have not been well-explored among interacting host species, is the relationships among the insects studied. Which parasitoid species attack which species of Drosophila in this community? Which parasitoid species are generalists vs specialists, what are the phylogenetic relationships among the various wasp and fly species collected, and how may these types of ecological and evolutionary relationships dictate what the authors expected to find regarding heritable viruses within each? I think a discussion of these dynamics would improve the manuscript, particularly in the Introduction and Discussion sections, as they are relevant to the goal of understanding what roles viruses play within interacting species.
Related to the point above, I think the paper could benefit from a more in-depth dialogue of what factors the authors hypothesized might shape the viral communities in these insects and why. It is briefly mentioned in the Discussion section that the results support the evolution of specificity for these heritable viruses that leads to their infection of a single host, but I think the authors’ a priori expectations should also be discussed in the Introduction to contextualize the goals of the study. For example, the authors mention the wealth of information we now have on the ways heritable bacteria impact insect biology, while we know relatively little on how viruses shape these same communities. I think pertinent information in the Introduction could therefore be to indicate what factors we know impact heritable bacterial communities in insects, which could be the basis for hypotheses of whether viruses follow similar trends.
In general, I think the Introduction could be more succinct in several places, especially if adding more context to the study system and the hypotheses that prompted this work. For example, the second and third paragraphs (starting on L9 and L23) could be condensed into a single paragraph, since they share a similar theme (i.e. we know little of heritable virus communities and their interactions). Similarly, introductory paragraphs 4 and 5 (starting on L38 and L58) could be condensed into one paragraph that summarizes briefly the work presented in this study.
Is it possible that insect species was the greatest structuring factor to virus communities, due in part to how many different species were included (n = 15) in the analysis and how few variations comprised the other factors tested (n = 2) (e.g. only two different locations, two different months, and two different years)? If more locations and/or more time points were sampled, would this increase the authors’ ability to accurately determine which factor(s) explain the distribution of viruses identified? If so, I think this should be addressed in the Discussion.
Minor Comments:
L76: Please elaborate here on what the climate differences exist between the two locations.
L81: Can the authors include more information on how these isofemale lines were maintained for two generations in the lab? Specifically, is it possible that horizontal transmission of these viruses could have occurred among individuals of the same line, or between parasitoids and D. melanogaster host flies during these two generations? Is the lab strain of D. melanogaster free of viruses or could these host flies be contributing to the viral communities that ultimately were sequenced from wasps?
L232: How was superparasitism between uninfected female wasps and infected male wasps ensured? If some proportion of fly larvae were initially infested with an uninfected female wasp egg but received no supernumerary wasp eggs laid by virus-infected wasp mothers, the resulting female wasp offspring would still be uninfected, due to no exposure to the virus during parasitism. This scenario, if not checked, could lead to an underestimation of horizontal virus transmission rates, which I think should be reported in the results.
Figures 1 and 2: I found these heatmaps quite difficult to understand initially, due to the minimal description of their contents in the figure legends and the manuscript text. I think more detailed information should be added to each figure legend, such as what the different clustering axes refer to. I also think a summary of the results conveyed by these maps should be included in the Results section, such as after their first mention on L279.
L686: Phasmaviridae is used in this sentence, although I believe the experiments were performed on the iflavirus, correct?
https://doi.org/10.24072/pci.evolbiol.100701.rev13