
BROQUET Thomas
- Roscoff biology station, CNRS, Roscoff, France
- Hybridization / Introgression, Population Genetics / Genomics, Sexual Selection, Speciation
- recommender
Recommendation: 1
Review: 1
Recommendation: 1
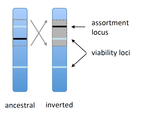
Prezygotic isolation, mating preferences, and the evolution of chromosomal inversions
The spread of chromosomal inversions as a mechanism for reinforcement
Recommended by Denis Roze and Thomas BroquetSeveral examples of chromosomal inversions carrying genes affecting mate choice have been reported from various organisms. Furthermore, inversions are also frequently involved in genetic isolation between populations or species. Past work has shown that inversions can spread when they capture not only some loci involved in mate choice but also loci involved in incompatibilities between hybridizing populations [1]. In this new paper [2], the authors derive analytical approximations for the selection coefficient associated with an inversion suppressing recombination between a locus involved in mate choice and one (or several) locus involved in Dobzhansky-Muller incompatibilities. Two mechanisms for mate choice are considered: assortative mating based on the allele present at a single locus, or a trait-preference model where one locus codes for the trait and another for the preference. The results show that such an inversion is generally favoured, the selective advantage associated with the inversion being strongest when hybridization is sufficiently frequent. Assuming pairwise epistatic interactions between loci involved in incompatibilities, selection for the inversion increases approximately linearly with the number of such loci captured by the inversion.
This paper is a good read for several reasons. First, it presents the problem clearly (e.g. the introduction provides a clear and concise presentation of the issue and past work) and its crystal-clear writing facilitates the reader's understanding of theoretical approaches and results. Second, the analysis is competently done and adds to previous work by showing that very general conditions are expected to be favourable to the spread of the type of inversion considered here. And third, it provides food for thought about the role of inversions in the origin or the reinforcement of divergence between nascent species. One result of this work is that an inversion linked to pre-zygotic isolation "is favoured so long as there is viability selection against recombinant genotypes", suggesting that genetic incompatibilities must have evolved first and that inversions capturing mating preference loci may then enhance pre-existing reproductive isolation. However, the results also show that inversions are more likely to be favoured in hybridizing populations among which gene flow is still high, rather than in more strongly isolated populations. This matches the observation that inversions are more frequently observed between sympatric species than between allopatric ones.
References
[1] Trickett AJ, Butlin RK. 1994. Recombination Suppressors and the Evolution of New Species. Heredity 73:339-345. doi: 10.1038/hdy.1994.180
[2] Dagilis AJ, Kirkpatrick M. 2016. Prezygotic isolation, mating preferences, and the evolution of chromosomal inversions. Evolution 70: 1465–1472. doi: 10.1111/evo.12954
Review: 1

Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands
Introgression from related species reveals fine-scale structure in an isolated population of mussels and causes patterns of genetic-environment associations
Recommended by Marianne Elias based on reviews by Thomas Broquet and Tatiana GiraudAssessing population connectivity is central to understanding population dynamics, and is therefore of great importance in evolutionary biology and conservation biology. In the marine realm, the apparent absence of physical barriers, large population sizes and high dispersal capacities of most organisms often result in no detectable structure, thereby hindering inferences of population connectivity. In a review paper, Gagnaire et al. [1] propose several ideas to improve detection of population connectivity. Notably, using simulations they show that under certain circumstances introgression from one species into another may reveal cryptic population structure within that second species.
The isolated Kerguelen archipelago in the south of Indian Ocean represents a typical situation where the structure of coastal marine organisms is expected to be difficult to detect. In an elegant genomic study, Fraïsse et al. [2] take advantage of introgression from foreign lineages to infer fine-grained population structure in a population of mussels around the Kerguelen archipelago, and investigate its association with environmental variables. Using a large panel of genome-wide markers (GBS) and applying a range of methods that unravel patterns of divergence and gene flow among lineages, they first find that the Kerguelen population is highly admixed, with a major genetic background corresponding to the southern mussel lineage Mytilus platensis introgressed by three northern lineages. By selecting a panel of loci enriched in ancestry-informative SNPs (ie, SNPs highly differentiated among northern lineages) they then detect a fine-scale genetic structure around the Kerguelen archipelago, and identify a major connectivity break. They further show an associating between the genetic structure and environmental variables, particularly the presence of Macrocystis kelp, a marker of habitat exposure to waves (a feature repeatedly evidenced to be important for mussels). While such association pattern could lead to the interpretation that differentiated SNPs correspond to loci directly under selection or linked with such loci, and even be considered as support for adaptive introgression, Fraïsse et al. [2] convincingly show by performing simulations that the genetic-environment association detected can be entirely explained by dispersal barriers associated with environmental variables (habitat-associated connectivity). They also explain why the association is better detected by ancestry-informative SNPs as predicted by Gagnaire et al. [1]. In addition, intrinsic genetic incompatibilities, which reduce gene flow, tend to become trapped at ecotones due to ecological selection, even when loci causing genetic incompatibilities are unlinked with loci involved in adaption to local ecological conditions (Bierne et al. [3]’s coupling hypothesis), leading to correlations between environmental variables and loci not involved in local adaptation. Notably, in Fraïsse et al. [2]’s study, the association between the kelp and ancestry-informative alleles is not consistent throughout the archipelago, casting further doubt on the implication of these alleles in local adaptation.
The study of Fraïsse et al. [2] is therefore an important contribution to evolutionary biology because 1) it provides an empirical demonstration that alleles of foreign origin can be pivotal to detect fine-scale connectivity patterns and 2) it represents a test case of Bierne et al. [3]’s coupling hypothesis, whereby introgressed alleles also enhance patterns of genetic-environment associations. Since genomic scan or GWAS approaches fail to clearly reveal loci involved in local adaptation, how can we disentangle environment-driven selection from intrinsic reproductive barriers and habitat-associated connectivity? A related question is whether we can reliably identify cases of adaptive introgression, which have increasingly been put forward as a mechanism involved in adaptation [4]. Unfortunately, there is no easy answer, and the safest way to go is to rely – where possible – on independent information [5], in particular functional studies of the detected loci, as is for example the case in the mimetic butterfly Heliconius literature (e. g., [6]) where several loci controlling colour pattern variation are well characterized.
References
[1] Gagnaire, P.-A., Broquet, T., Aurelle, D., Viard, F., Souissi, A., Bonhomme, F., Arnaud-Haond, S., & Bierne, N. (2015). Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evolutionary Applications, 8, 769–786. doi: 10.1111/eva.12288
[2] Fraïsse, C., Haguenauer, A., Gerard, K., Weber, A. A.-T., Bierne, N., & Chenuil, A. (2018). Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands. bioRxiv, 239244, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/239244
[3] Bierne, N., Welch, J., Loire, E., Bonhomme, F., & David, P. (2011). The coupling hypothesis: why genome scans may fail to map local adaptation genes. Molecular Ecology, 20, 2044–2072. doi: 10.1111/j.1365-294X.2011.05080.x
[4] Hedrick, P. W. (2013). Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Molecular Ecology, 22, 4606–4618. doi: 10.1111/mec.12415
[5] Ravinet, M., Faria, R., Butlin, R. K., Galindo, J., Bierne, N., Rafajlović, M., Noor, M. A. F., Mehlig, B., & Westram, A. M. (2017). Interpreting the genomic landscape of speciation: a road map for finding barriers to gene flow. Journal of Evolutionary Biology, 30, 1450–1477. doi: 10.1111/jeb.13047.
[6] Jay, P., Whibley, A., Frézal, L., Rodríguez de Cara, M. A., Nowell, R. W., Mallet, J., Dasmahapatra, K. K., & Joron, M. (2018). Supergene evolution triggered by the introgression of a chromosomal inversion. Current Biology, 28, 1839–1845.e3. doi: 10.1016/j.cub.2018.04.072