
ACHAZ Guillaume
- ISYEB and CIRB, MNHN / Univ. Pierre et Marie Curie / Collège de France, Paris, France
- Adaptation, Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Theory, Genome Evolution, Macroevolution, Molecular Evolution, Other, Phylogenetics / Phylogenomics, Population Genetics / Genomics
- recommender
Recommendations: 3
Review: 1
Recommendations: 3
Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between
Molecular evolution through the joint lens of genomic and population processes.
Recommended by Guillaume Achaz based on reviews by Benoit Nabholz and 1 anonymous reviewerIn their perspective article, F Pouyet and KJ Gilbert (2020), propose an interesting overview of all the processes that sculpt patterns of molecular evolution. This well documented article covers most (if not all) important facets of the recurrent debate that has marked the history of molecular evolution: the relative importance of natural selection and neutral processes (i.e. genetic drift). I particularly enjoyed reading this review, that instead of taking a clear position on the debate, catalogs patiently every pieces of information that can help understand how patterns we observed at the genome level, can be understood from a selectionnist point of view, from a neutralist one, and, to quote their title, from "everything in between". The review covers the classical objects of interest in population genetics (genetic drift, selection, demography and structure) but also describes several genomic processes (meiotic drive, linked selection, gene conversion and mutation processes) that obscure the interpretation of these population processes. The interplay between all these processes is very complex (to say the least) and have resulted in many cases in profound confusions while analyzing data. It is always very hard to fully acknowledge our ignorance and we have many times payed the price of model misspecifications. This review has the grand merit to improve our awareness in many directions. Being able to cover so many aspects of a wide topic, while expressing them simply and clearly, connecting concepts and observations from distant fields, is an amazing "tour de force". I believe this article constitutes an excellent up-to-date introduction to the questions and problems at stake in the field of molecular evolution and will certainly also help established researchers by providing them a stimulating overview supported with many relevant references.
References
[1] Pouyet F, Gilbert KJ (2020) Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between. arXiv:1909.11490 [q-bio]. ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. url:https://arxiv.org/abs/1909.11490
Separate the wheat from the chaff: genomic analysis of local adaptation in the red coral Corallium rubrum
Pros and Cons of local adaptation scans
Recommended by Guillaume Achaz based on reviews by Lucas Gonçalves da Silva and 1 anonymous reviewerThe preprint by Pratlong et al. [1] is a well thought quest for genomic regions involved in local adaptation to depth in a species a red coral living the Mediterranean Sea. It first describes a pattern of structuration and then attempts to find candidate genes involved in local adaptation by contrasting deep with shallow populations. Although the pattern of structuration is clear and meaningful, the candidate genomic regions involved in local adaptation remain to be confirmed. Two external reviewers and myself found this preprint particularly interesting regarding the right-mindedness of the authors in front of the difficulties they encounter during their experiments. The discussions on the pros and cons of the approach are very sound and can be easily exported to a large number of studies that hunt for local adaptation. In this sense, the lessons one can learn by reading this well documented manuscript are certainly valuable for a wide range of evolutionary biologists.
More precisely, the authors RAD-sequenced 6 pairs of 'shallow vs deep' samples located in 3 geographical sea areas (Banyuls, Corsica and Marseilles). They were hoping to detect genes involved in the adaptation to depth, if there were any. They start by assessing the patterns of structuration of the 6 samples using PCA and AMOVA [2] and also applied the STRUCTURE [3] assignment software. They show clearly that the samples were mostly differentiated between geographical areas and that only 1 out the 3 areas shows a pattern of isolation by depth (i.e. Marseille). They nevertheless went on and scanned for variants that are highly differentiated in the deep samples when compared to the shallow paired samples in Marseilles, using an Fst outliers approach [4] implemented in the BayeScEnv software [5]. No clear functional signal was in the end detected among the highly differentiated SNPs, leaving a list of candidates begging for complementary data.
The scan for local adaptation using signatures of highly divergent regions is a classical problem of population genetics. It has been applied on many species with various degrees of success. This study is a beautiful example of a well-designed study that did not give full satisfactory answers. Readers will especially appreciate the honesty and the in-depth discussions of the authors while exposing their results and their conclusions step by step.
References
[1] Pratlong, M., Haguenauer, A., Brener, K., Mitta, G., Toulza, E., Garrabou, J., Bensoussan, N., Pontarotti P., & Aurelle, D. (2018). Separate the wheat from the chaff: genomic scan for local adaptation in the red coral Corallium rubrum. bioRxiv, 306456, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/306456
[2] Excoffier, L., Smouse, P. E. & Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics, 131(2), 479-491.
[3] Pritchard, J. K., Stephens, M., & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945-959.
[4] Lewontin, R. C., & Krakauer, J. (1973). Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics, 74(1), 175-195.
[5] de Villemereuil, P., & Gaggiotti, O. E. (2015). A new FST‐based method to uncover local adaptation using environmental variables. Methods in Ecology and Evolution, 6(11), 1248-1258. doi: 10.1111/2041-210X.12418
Convergent evolution as an indicator for selection during acute HIV-1 infection
Is convergence an evidence for positive selection?
Recommended by Guillaume Achaz based on reviews by Jeffrey Townsend and 1 anonymous reviewerThe preprint by Bertels et al. [1] reports an interesting application of the well-accepted idea that positively selected traits (here variants) can appear several times independently; think about the textbook examples of flight capacity. Hence, the authors assume that reciprocally convergence implies positive selection. The methodology becomes then, in principle, straightforward as one can simply count variants in independent datasets to detect convergent mutations.
In this preprint, the authors have applied this counting strategy on 95 available sequence alignments of the env gene of HIV-1 [2,3] that corresponds to samples taken in different patients during the early phase of infection, at the very beginning of the onset of the immune system. They have compared the number and nature of the convergent mutations to a "neutral" model that assumes (a) a uniform distribution of mutations and (b) a substitution matrix estimated from the data. They show that there is an excess of convergent mutations when compared to the “neutral” expectations, especially for mutations that have arisen in 4+ patients. They also show that the gp41 gene is enriched in these convergent mutations. The authors then discuss in length the potential artifacts that could have given rise to the observed pattern.
I think that this preprint is remarkable in the proposed methodology. Samples are taken in different individuals, whose viral populations were founded by a single particle. Thus, there is no need for phylogenetic reconstruction of ancestral states that is the typical first step of trait convergent analyses. It simply becomes counting variants. This simple counting procedure needs nonetheless to be compared to a “neutral” expectation (a reference model), which includes the mutational process. In this article, the poor predictions of a specifically designed reference model is interpreted as an evidence for positive selection.
Whether the few mutations that are convergent in 4-7 samples out of 95 were selected or not is hard to assess with certainty. The authors have provided good evidence that they are, but only experimental validations will strengthen the claim. Nonetheless, beyond a definitive clue to the implication of selection on these particular mutations, I found the methodological strategy and the discussions on the potential biases highly stimulating. This article is an excellent starting point for further methodological developments that could be then followed by large-scale analyses of convergence in many different organisms and case studies.
References
[1] Bertels, F., Metzner, K. J., & Regoes R. R. (2018). Convergent evolution as an indicator for selection during acute HIV-1 infection. BioRxiv, 168260, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/168260
[2] Keele, B. F., Giorgi, E. E., Salazar-Gonzalez, J. F., Decker, J. M., Pham, K.T., Salazar, M. G., Sun, C., Grayson, T., Wang, S., Li, H. et al. (2008). Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci USA 105: 7552–7557. doi: 10.1073/pnas.0802203105
[3] Li, H., Bar, K. J., Wang, S., Decker, J. M., Chen, Y., Sun, C., Salazar-Gonzalez, J.F., Salazar, M.G., Learn, G.H., Morgan, C. J. et al. (2010). High multiplicity infection by HIV-1 in men who have sex with men. PLoS Pathogens 6:e1000890. doi: 10.1371/journal.ppat.1000890
Review: 1
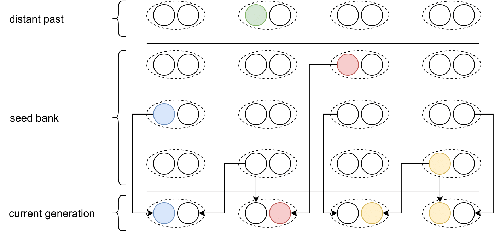
Weak seed banks influence the signature and detectability of selective sweeps
New insights into the dynamics of selective sweeps in seed-banked species
Recommended by Renaud Vitalis based on reviews by Guillaume Achaz, Jere Koskela, William Shoemaker and Simon BoitardMany organisms across the Tree of life have the ability to produce seeds, eggs, cysts, or spores, that can remain dormant for several generations before hatching. This widespread adaptive trait in bacteria, fungi, plants and animals, has a significant impact on the ecology, population dynamics and population genetics of species that express it (Evans and Dennehy 2005).
In population genetics, and despite the recognition of its evolutionary importance in many empirical studies, few theoretical models have been developed to characterize the evolutionary consequences of this trait on the level and distribution of neutral genetic diversity (see, e.g., Kaj et al. 2001; Vitalis et al. 2004), and also on the dynamics of selected alleles (see, e.g., Živković and Tellier 2018). However, due to the complexity of the interactions between evolutionary forces in the presence of dormancy, the fate of selected mutations in their genomic environment is not yet fully understood, even from the most recently developed models.
In a comprehensive article, Korfmann et al. (2023) aim to fill this gap by investigating the effect of germ banking on the probability of (and time to) fixation of beneficial mutations, as well as on the shape of the selective sweep in their vicinity. To this end, Korfmann et al. (2023) developed and released their own forward-in-time simulator of genome-wide data, including neutral and selected polymorphisms, that makes use of Kelleher et al.’s (2018) tree sequence toolkit to keep track of gene genealogies.
The originality of Korfmann et al.’s (2023) study is to provide new quantitative results for the effect of dormancy on the time to fixation of positively selected mutations, the shape of the genomic landscape in the vicinity of these mutations, and the temporal dynamics of selective sweeps. Their major finding is the prediction that germ banking creates narrower signatures of sweeps around positively selected sites, which are detectable for increased periods of time (as compared to a standard Wright-Fisher population).
The availability of Korfmann et al.’s (2023) code will allow a wider range of parameter values to be explored, to extend their results to the particularities of the biology of many species. However, as they chose to extend the haploid coalescent model of Kaj et al. (2001), further development is needed to confirm the robustness of their results with a more realistic diploid model of seed dormancy.
REFERENCES
Evans, M. E. K., and J. J. Dennehy (2005) Germ banking: bet-hedging and variable release from egg and seed dormancy. The Quarterly Review of Biology, 80(4): 431-451. https://doi.org/10.1086/498282
Kaj, I., S. Krone, and M. Lascoux (2001) Coalescent theory for seed bank models. Journal of Applied Probability, 38(2): 285-300. https://doi.org/10.1239/jap/996986745
Kelleher, J., K. R. Thornton, J. Ashander, and P. L. Ralph (2018) Efficient pedigree recording for fast population genetics simulation. PLoS Computational Biology, 14(11): e1006581. https://doi.org/10.1371/journal.pcbi.1006581
Korfmann, K., D. Abu Awad, and A. Tellier (2023) Weak seed banks influence the signature and detectability of selective sweeps. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.04.26.489499
Vitalis, R., S. Glémin, and I. Olivieri (2004) When genes go to sleep: the population genetic consequences of seed dormancy and monocarpic perenniality. American Naturalist, 163(2): 295-311. https://doi.org/10.1086/381041
Živković, D., and A. Tellier (2018). All but sleeping? Consequences of soil seed banks on neutral and selective diversity in plant species. Mathematical Modelling in Plant Biology, 195-212. https://doi.org/10.1007/978-3-319-99070-5_10