
RANNALA Bruce
- Department of Evolution and Ecology, UC Davis, Davis, United States of America
- Bioinformatics & Computational Biology, Evolutionary Theory, Human Evolution, Molecular Evolution, Other, Phylogenetics / Phylogenomics, Phylogeography & Biogeography, Population Genetics / Genomics, Speciation, Systematics / Taxonomy
- recommender
Recommendations: 2
Review: 1
Recommendations: 2
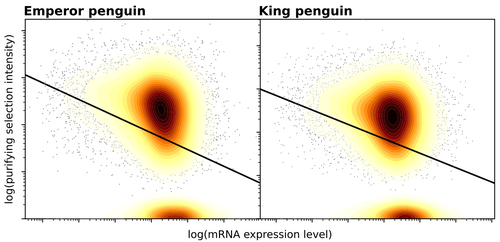
Gene expression is the main driver of purifying selection in large penguin populations
Purifying selection on highly expressed genes in Penguins
Recommended by Bruce Rannala based on reviews by Tanja Pyhäjärvi and 1 anonymous reviewerGiven the general importance of protein expression levels, in cells it is widely accepted that gene expression levels are often a target of natural selection and that most mutations affecting gene expression levels are therefore likely to be deleterious [1]. However, it is perhaps less obvious that the strength of selection on the regulated genes themselves may be influenced by their expression levels. This might be due to harmful effects of misfolded proteins, for example, when higher protein concentrations exist in cells [2]. Recent studies have suggested that highly expressed genes accumulate fewer deleterious mutations; thus a positive relationship appears to exist between gene expression levels and the relative strength of purifying selection [3].
The recommended paper by Trucchi et al. [4] examines the relationship between gene expression, purifying selection and a third variable -- effective population size -- in populations of two species of penguin with different population sizes, the Emperor penguin (Aptenodytes forsteri) and the King penguin (A. patagonicus). Using transcriptomic data and computer simulations modeling selection, they examine patterns of nonsynonymous and synonymous segregating polymorphisms (p) across genes in the two populations, concluding that even in relatively small populations purifying selection has an important effect in eliminating deleterious mutations.
References
1] Gilad Y, Oshlack A, and Rifkin SA. 2006. Natural selection on gene expression. Trends in Genetics 22: 456-461. https://doi.org/10.1016/j.tig.2006.06.002
[2] Yang JR, Liao BY, Zhuang SM, and Zhang J. 2012. Protein misinteraction avoidance causes highly expressed proteins to evolve slowly. Proceedings of the National Academy of Sciences 109: E831-E840. https://doi.org/10.1073/pnas.1117408109
[3] Duret L, and Mouchiroud D (2000). Determinants of substitution rates in mammalian genes: expression pattern affects selection intensity but not mutation rate. Molecular Biology and Evolution 17; 68-070. https://doi.org/10.1093/oxfordjournals.molbev.a026239
[4] Trucchi E, Massa P, Giannelli F, Latrille T, Fernandes FAN, Ancona L, Stenseth NC, Obiol JF, Paris J, Bertorelle G, and Le Bohec, C. 2023. Gene expression is the main driver of purifying selection in large penguin populations. bioRxiv 2023.08.08.552445, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.08.08.552445

Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic data
Predicting small ancestors using contemporary genomes of large mammals
Recommended by Bruce Rannala based on reviews by Bruce Rannala and 1 anonymous reviewerRecent methodological developments and increased genome sequencing efforts have introduced the tantalizing possibility of inferring ancestral phenotypes using DNA from contemporary species. One intriguing application of this idea is to exploit the apparent correlation between substitution rates and body size to infer ancestral species' body sizes using the inferred patterns of substitution rate variation among species lineages based on genomes of extant species [1].
The recommended paper by Figuet et al. [2] examines the utility of such approaches by analyzing the Cetartiodactyla, a clade of large mammals that have mostly well resolved phylogenetic relationships and a reasonably good fossil record. This combination of genomic data and fossils allows a direct comparison between body size predictions obtained from the genomic data and empirical evidence from the fossil record. If predictions seem good in groups such as the Cetartiodactyla, where there is independent evidence from the fossil record, this would increase the credibility of predictions made for species with less abundant fossils.
Figuet et al. [2] analyze transcriptome data for 41 species and report a significant effect of body mass on overall substitution rate, synonymous vs. non-synonymous rates, and the dynamics of GC-content, thus allowing a prediction of small ancestral body size in this group despite the fact that the extant species that were analyzed are nearly all large.
A comparative method based solely on morphology and phylogenetic relationships would be very unlikely to make such a prediction. There are many sources of uncertainty in the variables and parameters associated with these types of approaches: phylogenetic uncertainty (topology and branch lengths), uncertainty about inferred substitution rates, and so on. Although the authors do not account for all these sources of uncertainty the fact that their predicted body sizes appear sensible is encouraging and undoubtedly the methods will become more statistically sophisticated over time.
References
[1] Romiguier J, Ranwez V, Douzery EJP and Galtier N. 2013. Genomic evidence for large, long-lived ancestors to placental mammals. Molecular Biology and Evolution 30: 5–13. doi: 10.1093/molbev/mss211
[2] Figuet E, Ballenghien M, Lartillot N and Galtier N. 2017. Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic data. bioRxiv, ver. 3 of 4th December 2017. 139147. doi: 10.1101/139147
Review: 1
Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic data
Predicting small ancestors using contemporary genomes of large mammals
Recommended by Bruce Rannala based on reviews by Bruce Rannala and 1 anonymous reviewerRecent methodological developments and increased genome sequencing efforts have introduced the tantalizing possibility of inferring ancestral phenotypes using DNA from contemporary species. One intriguing application of this idea is to exploit the apparent correlation between substitution rates and body size to infer ancestral species' body sizes using the inferred patterns of substitution rate variation among species lineages based on genomes of extant species [1].
The recommended paper by Figuet et al. [2] examines the utility of such approaches by analyzing the Cetartiodactyla, a clade of large mammals that have mostly well resolved phylogenetic relationships and a reasonably good fossil record. This combination of genomic data and fossils allows a direct comparison between body size predictions obtained from the genomic data and empirical evidence from the fossil record. If predictions seem good in groups such as the Cetartiodactyla, where there is independent evidence from the fossil record, this would increase the credibility of predictions made for species with less abundant fossils.
Figuet et al. [2] analyze transcriptome data for 41 species and report a significant effect of body mass on overall substitution rate, synonymous vs. non-synonymous rates, and the dynamics of GC-content, thus allowing a prediction of small ancestral body size in this group despite the fact that the extant species that were analyzed are nearly all large.
A comparative method based solely on morphology and phylogenetic relationships would be very unlikely to make such a prediction. There are many sources of uncertainty in the variables and parameters associated with these types of approaches: phylogenetic uncertainty (topology and branch lengths), uncertainty about inferred substitution rates, and so on. Although the authors do not account for all these sources of uncertainty the fact that their predicted body sizes appear sensible is encouraging and undoubtedly the methods will become more statistically sophisticated over time.
References
[1] Romiguier J, Ranwez V, Douzery EJP and Galtier N. 2013. Genomic evidence for large, long-lived ancestors to placental mammals. Molecular Biology and Evolution 30: 5–13. doi: 10.1093/molbev/mss211
[2] Figuet E, Ballenghien M, Lartillot N and Galtier N. 2017. Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic data. bioRxiv, ver. 3 of 4th December 2017. 139147. doi: 10.1101/139147