
SHAPIRO B. Jesse
- Department of Microbiology & Immunology, McGill University, Montréal, Canada
- Bioinformatics & Computational Biology, Evolutionary Ecology, Genome Evolution, Molecular Evolution, Phylogenetics / Phylogenomics, Population Genetics / Genomics, Speciation
- recommender
Recommendations: 2
Reviews: 0
Recommendations: 2
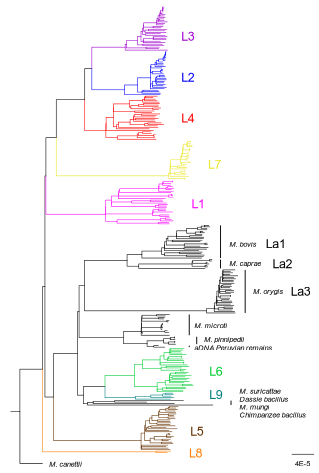
How do monomorphic bacteria evolve? The Mycobacterium tuberculosis complex and the awkward population genetics of extreme clonality
How the tubercle bacillus got its genome: modernising, modelling, and making sense of the stories we tell
Recommended by B. Jesse Shapiro based on reviews by 2 anonymous reviewersIn this instructive review, Stritt and Gagneux offer a balanced perspective on the evolutionary forces shaping Mycobacterium tuberculosis and make the case that our instinct for storytelling be balanced with quantitative models. M. tuberculosis is perhaps the best-known clonal bacterial pathogen – evolving largely in the absence of horizontal gene transfer. Its genome is full of puzzling patterns, including much higher GC content than most intracellular pathogens (which suggests efficient selection to resist AT-skewed mutational bias) but a very high ratio of nonsynonymous to synonymous substitution rates (dN/dS ~ 0.5, typically interpreted as weak selection against deleterious amino acid changes).
The authors offer alternative explanations for these patterns, framing the question: is M. tuberculosis evolution shaped mainly by drift or by efficient selection? They propose that this question can only be answered by accounting for the pathogen’s extreme clonality. A clonal lifestyle can have its advantages, for example when adaptations must arise in a particular order (Kondrashov and Kondrashov 2001). An important disadvantage highlighted by the authors are linkage effects: without recombination to shuffle them up, beneficial mutations are linked to deleterious mutations in the same genome (hitchhiking) and purging deleterious mutations also purges neutral diversity across the genome (background selection). The authors propose the latter – efficient purifying selection and strong linkage – as an explanation for the low genetic diversity observed in M. tuberculosis. This is of course not exclusive of other related explanations, such as clonal interference (Gerrish and Lenski 1998). They also champion the use of forward evolutionary simulations (Haller and Messer 2019) to model the interplay between selection, recombination, and demography as a powerful alternative to traditional backward coalescent models.
At times, Stritt and Gagneux are pessimistic about our existing methods – arguing that dN/dS and homoplasies “tell us little about the frequency and strength of selection.” Even though I favour a more optimistic view, I fully agree that our traditional population genetic metrics are sensitive to a slew of different deviations from a standard neutral evolution model and must be interpreted with caution. As I and others have argued, the extent of recombination (measured as the amount of linkage in a genome) is a key factor in determining how best to test for natural selection (Shapiro et al. 2009) and to conduct genotype-phenotype association studies (Chen and Shapiro 2021) in microbes. While this article is focused on the well-studied M. tuberculosis complex, there are many parallels with other clonal bacteria, including pathogens and symbionts. Whatever your favourite bug, we must all be careful to make sure the stories we tell about them are not “just so tales” but are supported, to the extent possible, by data and quantitative models.
References
Chen, Peter E., and B. Jesse Shapiro. 2021. "Classic Genome-Wide Association Methods Are Unlikely to Identify Causal Variants in Strongly Clonal Microbial Populations." bioRxiv.
https://doi.org/10.1101/2021.06.30.450606
Gerrish, P. J., and R. E. Lenski. 1998. "The Fate of Competing Beneficial Mutations in an Asexual Population." Genetica 102-103 (1-6): 127-44.
https://doi.org/10.1023/A:1017067816551
Haller, Benjamin C., and Philipp W. Messer. 2019. "SLiM 3: Forward Genetic Simulations Beyond the Wright-Fisher Model." Molecular Biology and Evolution 36 (3): 632-37.
https://doi.org/10.1093/molbev/msy228
Kondrashov, F. A., and A. S. Kondrashov. 2001. "Multidimensional Epistasis and the Disadvantage of Sex." Proceedings of the National Academy of Sciences of the United States of America 98 (21): 12089-92.
https://doi.org/10.1073/pnas.211214298
Shapiro, B. Jesse, Lawrence A. David, Jonathan Friedman, and Eric J. Alm. 2009. "Looking for Darwin's Footprints in the Microbial World." Trends in Microbiology 17 (5): 196-204.
https://doi.org/10.1016/j.tim.2009.02.002
Stritt, C., Gagneux, S. (2023). How do monomorphic bacteria evolve? The Mycobacterium tuberculosis complex and the awkward population genetics of extreme clonality. EcoEvoRxiv, ver.3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.32942/X2GW2P
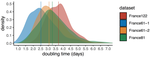
Early phylodynamics analysis of the COVID-19 epidemics in France
SARS-Cov-2 genome sequence analysis suggests rapid spread followed by epidemic slowdown in France
Recommended by B. Jesse Shapiro based on reviews by Luca Ferretti and 2 anonymous reviewersSequencing and analyzing SARS-Cov-2 genomes in nearly real time has the potential to quickly confirm (and inform) our knowledge of, and response to, the current pandemic [1,2]. In this manuscript [3], Danesh and colleagues use the earliest set of available SARS-Cov-2 genome sequences available from France to make inferences about the timing of the major epidemic wave, the duration of infections, and the efficacy of lockdown measures. Their phylodynamic estimates -- based on fitting genomic data to molecular clock and transmission models -- are reassuringly close to estimates based on 'traditional' epidemiological methods: the French epidemic likely began in mid-January or early February 2020, and spread relatively rapidly (doubling every 3-5 days), with people remaining infectious for a median of 5 days [4,5]. These transmission parameters are broadly in line with estimates from China [6,7], but are currently unknown in France (in the absence of contact tracing data). By estimating the temporal reproductive number (Rt), the authors detected a slowing down of the epidemic in the most recent period of the study, after mid-March, supporting the efficacy of lockdown measures.
Along with the three other reviewers of this manuscript, I was impressed with the careful and exhaustive phylodynamic analyses reported by Danesh et al. [3]. Notably, they take care to show that the major results are robust to the choice of priors and to sampling. The authors are also careful to note that the results are based on a limited sample size of SARS-Cov-2 genomes, which may not be representative of all regions in France. Their analysis also focused on the dominant SARS-Cov-2 lineage circulating in France, which is also circulating in other countries. The variations they inferred in epidemic growth in France could therefore be reflective on broader control policies in Europe, not only those in France. Clearly more work is needed to fully unravel which control policies (and where) were most effective in slowing the spread of SARS-Cov-2, but Danesh et al. [3] set a solid foundation to build upon with more data. Overall this is an exemplary study, enabled by rapid and open sharing of sequencing data, which provides a template to be replicated and expanded in other countries and regions as they deal with their own localized instances of this pandemic.
References
[1] Grubaugh, N. D., Ladner, J. T., Lemey, P., Pybus, O. G., Rambaut, A., Holmes, E. C., & Andersen, K. G. (2019). Tracking virus outbreaks in the twenty-first century. Nature microbiology, 4(1), 10-19. doi: 10.1038/s41564-018-0296-2
[2] Fauver et al. (2020) Coast-to-Coast Spread of SARS-CoV-2 during the Early Epidemic in the United States. Cell, 181(5), 990-996.e5. doi: 10.1016/j.cell.2020.04.021
[3] Danesh, G., Elie, B., Michalakis, Y., Sofonea, M. T., Bal, A., Behillil, S., Destras, G., Boutolleau, D., Burrel, S., Marcelin, A.-G., Plantier, J.-C., Thibault, V., Simon-Loriere, E., van der Werf, S., Lina, B., Josset, L., Enouf, V. and Alizon, S. and the COVID SMIT PSL group (2020) Early phylodynamics analysis of the COVID-19 epidemic in France. medRxiv, 2020.06.03.20119925, ver. 3 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/2020.06.03.20119925
[4] Salje et al. (2020) Estimating the burden of SARS-CoV-2 in France. hal-pasteur.archives-ouvertes.fr/pasteur-02548181
[5] Sofonea, M. T., Reyné, B., Elie, B., Djidjou-Demasse, R., Selinger, C., Michalakis, Y. and Samuel Alizon, S. (2020) Epidemiological monitoring and control perspectives: application of a parsimonious modelling framework to the COVID-19 dynamics in France. medRxiv, 2020.05.22.20110593. doi: 10.1101/2020.05.22.20110593
[6] Rambaut, A. (2020) Phylogenetic analysis of nCoV-2019 genomes. virological.org/t/phylodynamic-analysis-176-genomes-6-mar-2020/356
[7] Li et al. (2020) Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N Engl J Med, 382: 1199-1207. doi: 10.1056/NEJMoa2001316