
GALTIER Nicolas
- Institut des Sciences de l'Evolution, CNRS - Université Montpellier - IRD - EPHE, Montpellier, France
- Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Phylogenetics / Phylogenomics, Population Genetics / Genomics
- recommender
Recommendations: 4
Reviews: 0
Recommendations: 4
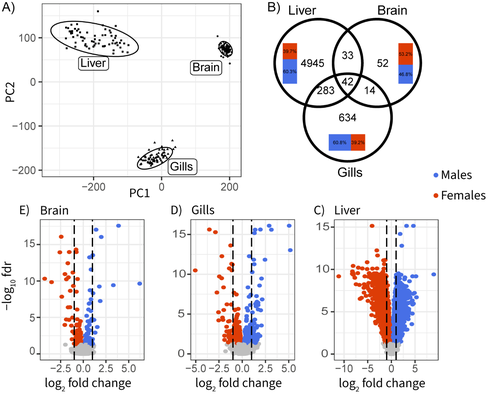
Sex-biased gene expression across tissues reveals unexpected differentiation in the gills of the threespine stickleback
Sex-biased gene expression in fish – a milestone
Recommended by Nicolas Galtier based on reviews by Qi Zhou and 2 anonymous reviewersThere is a heavy body of literature on sex-biased gene expression, which can easily be tricky. One reason is that expression data are multi-dimensional, notoriously noisy, and highly sensitive to experimental conditions. Achieving reproducibility is, therefore, a challenge, especially in non-model organisms. Another reason is that the evolutionary forces shaping gene expression variation are complex, involving processes such as intra- and inter-locus conflicts between sexes, sex-chromosome evolution and degeneration, dosage compensation, cis vs. trans regulation, dominance, linkage, drift, etc... Not surprisingly, the field is rich in discordant studies, and a wide variety of situations have been described.
The three-spined stickleback is a good illustration. This widely-studied fish displays conspicuous sexual dimorphisms, both morphological and behavioural. Yet, the existing literature regarding which genes and tissues are involved is remarkably unclear due to the lack of a comprehensive data set. Sylvestre et al. (1) here fix this issue. They sampled 40 wild-caught individuals in each of the two sexes and performed high-throughput sequencing of the transcriptome in three somatic tissues, dissected and preserved in the exact same conditions. This is an impressive effort, well above current standards. Data analysis delivered a series of neat results: gene expression in the liver is particularly sex-biased; the brain, in contrast, is remarkably little sex-differentiated despite the presence of courtship and paternal care in this species; gills show significantly sex-biased gene expression, which had been unnoticed previously despite the importance of this organ in fish ecotoxicology; the relatively young sex-chromosomes, finally, do not seem to experience dosage compensation, and are therefore enriched in sex-biased genes.
Some of these results are consistent with previous studies in other fish species (2), here confirmed or demonstrated with a high degree of certainty. Others are new and worth considering in future studies of stickleback ecology and reproduction. We simply need more studies of this sort: well-conducted and clear-cut, recalling the career of its last author.
References
(1) Florent Sylvestre, Nadia Aubin-Horth, Louis Bernatchez (2024) Sex-biased gene expression across tissues reveals unexpected differentiation in the gills of the threespine stickleback. bioRxiv, ver.2 peer-reviewed and recommended by PCI Evol Biol https://doi.org/10.1101/2024.06.09.597944
(2) Iulia Darolti I, Judith E. Mank (2023). Sex-biased gene expression at single-cell resolution: cause and consequence of sexual dimorphism. Evolution Letters 7(3):148-156. https://doi.org/10.1093/evlett/qrad013
Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pest
The scandalous pest
Recommended by Nicolas Galtier based on reviews by 2 anonymous reviewersKoutsovoulos et al. [1] have generated and analysed the first population genomic dataset in root-knot nematode Meloidogyne incognita. Why is this interesting? For two major reasons. First, M. incognita has been documented to be apomictic, i.e., to lack any form of sex. This is a trait of major evolutionary importance, with implications on species adaptive potential. The study of genome evolution in asexuals is fascinating and has the potential to inform on the forces governing the evolution of sex and recombination. Even small amounts of sex, however, are sufficient to restore most of the population genetic properties of true sexuals [2]. Because rare events of sex can remain undetected in the field, to confirm asexuality in M. incognita using genomic data is an important step. The second reason why M. incognita is of interest is that this nematode is one of the most harmful pests currently living on earth. M. incognita feeds on the roots of many cultivated plants, including tomato, bean, and cotton, and has been of major agricultural importance for decades. A number of races were defined based on host specificity. These have played a key role in attempts to control the dynamic of M. incognita populations via crop rotations. Races and management strategies so far lack any genetic basis, hence the second major interest of this study.
The authors newly sequenced the full genome of eleven strains from Brazil and added nine already available samples from Africa and North-America. They report that, in all likelihood, M. incognita is indeed a purely asexual species. This is supported by (i) the confirmation that the genome is in its major part haploid, and (ii) a spectacularly high level of linkage disequilibrium, which does not decline with genetic distance between loci at a 100kb scale. The absence of sex and recombination is associated in M. incognita with a remarkably low amount of genetic diversity - one order of magnitude less than in typical sexual nematodes - and an heavy load of deleterious mutations, as measured by the ratio of non-synonymous (=amino-acid changing) to synonymous (=amino-acid conservative) diversity in coding sequences. The other important result of this study is that the population substructure in M. incognita is in no way related to host races or geography. The tree genetic clusters that are identified include strains from several continents and feeding on a diversity of host plants.
The implications of this work are numerous. First, the results suggest that M. incognita is an ancient asexual. Asexuality, which was here demonstrated via linkage disequilibrium analysis, must be ancient enough for diploidy (or, in this case, maybe triploidy) to have been lost - i.e., formerly homologous chromosomes have accumulated enough mutations to be assembled as distinct entities. So we are not talking about a highly successful clone having recently spread the world - rather a long-term obligate parthenogen. Asexual organisms are deprived of the source of genetic variation offered by recombination, which is why asexuality is thought to be an evolutionary dead-end. Long-term asexuals are uncommon and even the most famous ones, bdelloid rotifers, are suspected to experience between-individual genetic transfers [3]. M. incognita is apparently a true 'evolutionary scandal', and as such deserves particular attention from molecular evolutionary geneticists.
The lack of any host race effect on the genetic diversity of M. incognita is another important finding. So-called 'races' have largely contributed to shape researchers' view of the structure of the species so far. This study demonstrates that a mental effort is now needed to forget about races, and consider host-specificity for what it is - a phenotypic trait. This result implies that many host shifts must have independently occurred in the three M. incognita genetic lineages, suggesting an arms race between plants and nematodes, which in the absence of sex and recombination must be entirely mutation-driven on the nematode side. Genes functionally involved in the arms race might therefore be expected to have experienced convergent evolution, if distinct M. incognita lineages have adopted the same solutions to overcome plant defenses. The present study paves the way for such a genome scan. The authors rightly discuss that the strong adaptive potential of M. incognita, at least in terms of host shift, despite no sex and tiny amounts of genetic diversity, is a paradox that would deserve to be further investigated.
References
[1] Koutsovoulos, G. D., Marques, E., Arguel, M. J., Duret, L., Machado, A. C. Z., Carneiro, R. M. D. G., Kozlowski, D. K., Bailly-Bechet, M., Castagnone-Sereno, P., Albuquerque, E. V., & Danchin, E. G. J. (2019). Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pest. bioRxiv, 362129, ver. 5, peer-reviewed and recommended by Peer Community in Evolutionary Biology. doi: 10.1101/362129
[2] Hartfield, M. (2016). Evolutionary genetic consequences of facultative sex and outcrossing. Journal of evolutionary biology, 29(1), 5-22. doi: 10.1111/jeb.12770
[3] Debortoli, N., Li, X., Eyres, I., Fontaneto, D., Hespeels, B., Tang, C. Q., Flot, J. F. & Van Doninck, K. (2016). Genetic exchange among bdelloid rotifers is more likely due to horizontal gene transfer than to meiotic sex. Current Biology, 26(6), 723-732. doi: 10.1016/j.cub.2016.01.031
The quiescent X, the replicative Y and the Autosomes
Replication-independent mutations: a universal signature ?
Recommended by Nicolas Galtier based on reviews by Marc Robinson-Rechavi and Robert LanfearMutations are the primary source of genetic variation, and there is an obvious interest in characterizing and understanding the processes by which they appear. One particularly important question is the relative abundance, and nature, of replication-dependent and replication-independent mutations - the former arise as cells replicate due to DNA polymerization errors, whereas the latter are unrelated to the cell cycle. A recent experimental study in fission yeast identified a signature of mutations in quiescent (=non-replicating) cells: the spectrum of such mutations is characterized by an enrichment in insertions and deletions (indels) compared to point mutations, and an enrichment of deletions compared to insertions [2].
What Achaz et al. [1] report here is that the very same signature is detectable in humans. This time the approach is indirect and relies on two key aspects of mammalian reproduction biology: (1) oocytes remain quiescent over most of a female's lifespan, whereas spermatocytes keep dividing after male puberty, and (2) X chromosome, Y chromosome and autosomes spend different amounts of time in a female vs. male context. In agreement with the yeast study, Achaz et al. show that in humans the male-associated Y chromosome, for which quiescence is minimal, has by far the lowest ratios of indels to point mutations and of deletions to insertions, whereas the female-associated X chromosome has the highest. This is true both of variants that are polymorphic among humans and of fixed differences between humans and chimpanzees.
So we appear to be here learning about an important and general aspect of the mutation process. The authors suggest that, to a large extent, chromosomes tend to break in pieces at a rate that is proportional to absolute time - because indels in quiescent stage presumably result from double-strand DNA breaks. A very recent analysis of numerous mother-father-child trios in humans confirms this prediction in demonstrating an effect of maternal age, but not of paternal age, on the recombination rate [3]. This result also has important implications with respect to the interpretation of substitution rate variation among taxa and genomic compartments, particularly mitochondrial vs. nuclear, and their relationship with the generation time and longevity of organisms (e.g. [4]).
References
[1] Achaz, G., Gangloff, S., and Arcangioli, B. (2019). The quiescent X, the replicative Y and the Autosomes. BioRxiv, 351288, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/351288
[2] Gangloff, S., Achaz, G., Francesconi, S., Villain, A., Miled, S., Denis, C., and Arcangioli, B. (2017). Quiescence unveils a novel mutational force in fission yeast. eLife, 6:e27469. doi: 10.7554/eLife.27469
[3] Halldorsson, B. V., Palsson, G., Stefansson, O. A., Jonsson, H., Hardarson, M. T., Eggertsson, H. P., … Stefansson, K. (2019). Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science, 363: eaau1043. doi: 10.1126/science.aau1043
[4] Saclier, N., François, C. M., Konecny-Dupré, L., Lartillot, N., Guéguen, L., Duret, L., … Lefébure, T. (2019). Life History Traits Impact the Nuclear Rate of Substitution but Not the Mitochondrial Rate in Isopods. Molecular Biology and Evolution, in press. doi: 10.1093/molbev/msy247

Rates of Molecular Evolution Suggest Natural History of Life History Traits and a Post-K-Pg Nocturnal Bottleneck of Placentals
A new approach to DNA-aided ancestral trait reconstruction in mammals
Recommended by Nicolas Galtier and Belinda ChangReconstructing ancestral character states is an exciting but difficult problem. The fossil record carries a great deal of information, but it is incomplete and not always easy to connect to data from modern species. Alternatively, ancestral states can be estimated by modelling trait evolution across a phylogeny, and fitting to values observed in extant species. This approach, however, is heavily dependent on the underlying assumptions, and typically results in wide confidence intervals.
An alternative approach is to gain information on ancestral character states from DNA sequence data. This can be done directly when the trait of interest is known to be determined by a single, or a small number, of major effect genes. In some of these cases it can even be possible to investigate an ancestral trait of interest by inferring and resurrecting ancestral sequences in the laboratory. Examples where this has been successfully used to address evolutionary questions range from the nocturnality of early mammals [1], to the loss of functional uricases in primates, leading to high rates of gout, obesity and hypertension in present day humans [2]. Another possibility is to rely on correlations between species traits and the genome average substitution rate/process. For instance, it is well established that the ratio of nonsynonymous to synonymous substitution rate, dN/dS, is generally higher in large than in small species of mammals, presumably due to a reduced effective population size in the former. By estimating ancestral dN/dS, one can therefore gain information on ancestral body mass (e.g. [3-4]).
The interesting paper by Wu et al. [5] further develops this second possibility of incorporating information on rate variation derived from genomic data in the estimation of ancestral traits. The authors analyse a large set of 1185 genes in 89 species of mammals, without any prior information on gene function. The substitution rate is estimated for each gene and each branch of the mammalian tree, and taken as an indicator of the selective constraint applying to a specific gene in a specific lineage – more constraint, slower evolution. Rate variation is modelled as resulting from a gene effect, a branch effect, and a gene X branch interaction effect, which captures lineage-specific peculiarities in the distribution of functional constraint across genes. The interaction term in terminal branches is regressed to observed trait values, and the relationship is used to predict ancestral traits from interaction terms in internal branches. The power and accuracy of the estimates are convincingly assessed via cross validation. Using this method, the authors were also able to use an unbiased approach to determine which genes were the main contributors to the evolution of the life-history traits they reconstructed.
The ancestors to current placental mammals are predicted to have been insectivorous - meaning that the estimated distribution of selective constraint across genes in basal branches of the tree resembles that of extant insectivorous taxa - consistent with the mainstream palaeontological hypothesis. Another interesting result is the prediction that only nocturnal lineages have passed the Cretaceous/Tertiary boundary, so that the ancestors of current orders of placentals would all have been nocturnal. This suggests that the so-called "nocturnal bottleneck hypothesis" should probably be amended. Similar reconstructions are achieved for seasonality, sociality and monogamy – with variable levels of uncertainty.
The beauty of the approach is to analyse the variance, not only the mean, of substitution rate across genes, and their methods allow for the identification of the genes contributing to trait evolution without relying on functional annotations. This paper only analyses discrete traits, but the framework can probably be extended to continuous traits as well.
References
[1] Bickelmann C, Morrow JM, Du J, Schott RK, van Hazel I, Lim S, Müller J, Chang BSW, 2015. The molecular origin and evolution of dim-light vision in mammals. Evolution 69: 2995-3003. doi: https://doi.org/10.1111/evo.12794
[2] Kratzer, JT, Lanaspa MA, Murphy MN, Cicerchi C, Graves CL, Tipton PA, Ortlund EA, Johnson RJ, Gaucher EA, 2014. Evolutionary history and metabolic insights of ancient mammalian uricases. Proceedings of the National Academy of Science, USA 111:3763-3768. doi: https://doi.org/10.1073/pnas.1320393111
[3] Lartillot N, Delsuc F. 2012. Joint reconstruction of divergence times and life-history evolution in placental mammals using a phylogenetic covariance model. Evolution 66:1773-1787. doi: https://doi.org/10.1111/j.1558-5646.2011.01558.x
[4] Romiguier J, Ranwez V, Douzery EJ, Galtier N. 2013. Genomic evidence for large, long-lived ancestors to placental mammals. Molecular Biology and Evolution 30:5-13. doi: https://doi.org/10.1093/molbev/mss211
[5] Wu J, Yonezawa T, Kishino H. 2016. Rates of Molecular Evolution Suggest Natural History of Life History Traits and a Post-K-Pg Nocturnal Bottleneck of Placentals. Current Biology 27: 3025-3033. doi: https://doi.org/10.1016/j.cub.2017.08.043