
Simulating bacterial evolution forward-in-time
Simulation of bacterial populations with SLiM
Abstract
Recommendation: posted 15 February 2021, validated 04 March 2021
Bertels, F. (2021) Simulating bacterial evolution forward-in-time. Peer Community in Evolutionary Biology, 100123. https://doi.org/10.24072/pci.evolbiol.100123
Recommendation
Jean Cury and colleagues (2021) have developed a protocol to simulate bacterial evolution in SLiM. In contrast to existing methods that depend on the coalescent, SLiM simulates evolution forward in time. SLiM has, up to now, mostly been used to simulate the evolution of eukaryotes (Haller and Messer 2019), but has been adapted here to simulate evolution in bacteria. Forward-in-time simulations are usually computationally very costly. To circumvent this issue, bacterial population sizes are scaled down. One would now expect results to become inaccurate, however, Cury et al. show that scaled-down forwards simulations provide very accurate results (similar to those provided by coalescent simulators) that are consistent with theoretical expectations. Simulations were analyzed and compared to existing methods in simple and slightly more complex scenarios where recombination affects evolution. In all scenarios, simulation results from coalescent methods (fastSimBac (De Maio and Wilson 2017), ms (Hudson 2002)) and scaled-down forwards simulations were very similar, which is very good news indeed.
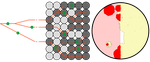
A biologist not aware of the complexities of forwards, backwards simulations and the coalescent, might now naïvely ask why another simulation method is needed if existing methods perform just as well. To address this question the manuscript closes with a very neat example of what exactly is possible with forwards simulations that cannot be achieved using existing methods. The situation modeled is the growth and evolution of a set of 50 bacteria that are randomly distributed on a petri dish. One side of the petri dish is covered in an antibiotic the other is antibiotic-free. Over time, the bacteria grow and acquire antibiotic resistance mutations until the entire artificial petri dish is covered with a bacterial lawn. This simulation demonstrates that it is possible to simulate extremely complex (e.g. real world) scenarios to, for example, assess whether certain phenomena are expected with our current understanding of bacterial evolution, or whether there are additional forces that need to be taken into account. Hence, forwards simulators could significantly help us to understand what current models can and cannot explain in evolutionary biology.
References
Cury J, Haller BC, Achaz G, Jay F (2021) Simulation of bacterial populations with SLiM. bioRxiv, 2020.09.28.316869, version 5 peer-reviewed and recommended by Peer community in Evolutionary Biology. https://doi.org/10.1101/2020.09.28.316869
De Maio N, Wilson DJ (2017) The Bacterial Sequential Markov Coalescent. Genetics, 206, 333–343. https://doi.org/10.1534/genetics.116.198796
Haller BC, Messer PW (2019) SLiM 3: Forward Genetic Simulations Beyond the Wright–Fisher Model. Molecular Biology and Evolution, 36, 632–637. https://doi.org/10.1093/molbev/msy228
Hudson RR (2002) Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics, 18, 337–338. https://doi.org/10.1093/bioinformatics/18.2.337

The recommender in charge of the evaluation of the article and the reviewers declared that they have no conflict of interest (as defined in the code of conduct of PCI) with the authors or with the content of the article. The authors declared that they comply with the PCI rule of having no financial conflicts of interest in relation to the content of the article.
Evaluation round #3
DOI or URL of the preprint: 10.1101/2020.09.28.316869
Version of the preprint: 3
Author's Reply, 10 Feb 2021
Decision by Frederic Bertels, posted 11 Jan 2021
Dear authors,
please revise your manuscript according to the reviewers' comments. Please make sure to address and reply to every comment.
Reviewed by anonymous reviewer 1, 06 Jan 2021
I want to thank the authors for improving a lot their manuscript. I highly appreciate your effort.
I hope you agree that your second version is more readable and precise than the first one.
In my case, almost all the comments I had have been answered.
-You changed the abstract
- You showcase an example
- you clarify gene conversion term
- the importance of considering a circular genome etc...
Two major suggestions:
However, I would like to suggest focusing on two critical aspects of the paper:
1. the figure legends are better, but they do not directly convey the entire message.
For example when I see Figure 1 I am not sure if rescaling factors have also been applied in FastSimBac and ms.
Would be a big plus to include a summary table of your results.
- It's a bit of a pain to read through the supplementary figures. Low quality, tiny labels. It's not easy to go through them.
One minor suggestion:
3. This is a minor suggestion: when you show the code on page 6 and 7 avoid better this format.
Use a grey shaded area similar to StackOverflow when you present a code case, so as the reader can copy paste and test the
code easily. That's an aesthetic suggestion.
In any case,
Thank you for your effort.
Happy new year
Reviewed by anonymous reviewer 2, 18 Dec 2020
I have taken a look at the authors' responses to my original comments and found them sound.
https://doi.org/10.24072/pci.evolbiol.100356.rev32Reviewed by anonymous reviewer 3, 06 Jan 2021
This revised manuscript is partly incorporating the reviewer's suggestions. Most of my points were taken care of, however, I see room for improvement for the description of the simulation, the clarity of the figure, and the explanation of the burn-in phase. My largest remaining concern is the discussion of selected recombination events. Here are my detailed points:
An overview figure is provided, however it is only in the supplement and it does not visualise the simulation parameters (Ne, generations, rho, tractlen, genomesize, hgtrate).
An additional simulation of bacteria under antibiotics on a Petri dish is now included. The choice of parameters is quite arbitrary, e.g., the antibiotic is reducing the fitness only to 0.47, I guess lower values are more realistic. However, the simulation should simply display an example of application and it serves this purpose. Nevertheless, method's details are still missing. How does the spatial model work, how is the neighbourhood of a bacterium defined? I guess this is a standard model, so references would help here a lot. I am also missing the information on recombination rate and tract length for this simulation.
I had provided several references for the discussion of realistic recombination tract lengths. Nevertheless, the authors decided to not discuss the range of recombination tract lengths in the manuscript and point to their simulations of length 1220bp 12,200bp and 122,000bp. I had also pointed out that their initial choice of a recombination tract length of 122kbp is based on "selected" recombination events. Thus, this estimate is not a good choice for simulations which should be based on unselected events. The authors ignored this point in their answer. I understand, that at this point of the manuscript it is not feasible to repeat the simulations, but the discussion of unselected vs. selected recombination events should at least be mentioned in the discussion. Otherwise the reference to the S. agalactiae length is misleading.
The authors added more information on the burn-in phase and also display it in the figure. As I understand, in the WF model, the burn-in has to be run before the SLiM simulation, however, with the nonWF model it is possible to run it afterwards only on the individuals that have descendants (as displayed in Fig. 8A). However, how can SLiM be run with selection if the diversity and the mutations are not yet clear at the start of the simulation? How is the fitness of the individuals known? I think I am missing a piece of information here.
https://doi.org/10.24072/pci.evolbiol.100356.rev33Evaluation round #2
DOI or URL of the preprint: 10.1101/2020.09.28.316869
Version of the preprint: 2
Author's Reply, 08 Dec 2020
Dear managing board members,
First of all we would like to thank everyone, board members, recommenders and reviewers for their work in general and for the time spent on our manuscript. On Monday November, 30th, we sent our answers to the reviewers’ comment along with an updated version of our manuscript “Simulation of bacterial population with SLiM”. About 14 hours later, we receive a notification from Peer Community In Evolutionary Biology, informing us that our paper would not be recommended. To our surprise, the recommender did not send our updated paper and replies to the reviewers and categorically rejected our paper based on critics that appear unfair to us. Hence, we would like to appeal this decision for the following reasons. The main reason for the recommender to reject our paper appears to be that we do not demonstrate that using SLiM does open new simulation possibilities, and to do so, we should, in addition to our paper, “simply solve an interesting biological problem”. The recommender adds “The reviewers and I feel like the value of the paper really hangs on the detailed analysis of such an example”. First we added this example, showcasing new simulation possibilities, as asked by a reviewer, who even suggested to add it in the discussion. Second we doubt that the reviewers saw this new example since the paper was not sent for another round of review. We do not intend to take the paper in that direction, because our study belongs to a “method and software” category and it not our intention to turn it into a “novel biological insights” type of manuscript by deepening the analysis of specific cases. Given PCI’s policy about the scope of a paper (”No need to examine whether the article falls within the scope of the PCI. Once a submission has been validated by the managing board, it is considered suitable for the PCI”), we do not understand the request of including a novel and detailed analysis of an “interesting biological problem". Besides, PCI Evol Biol precises that “Studies of methodologies for evolutionary biology are also appreciated”. Moreover, it is very solidly established and uncontroversial to say that forward simulation allows many important types of models to be run that are analytically intractable and cannot be simulated with the coalescent. This is the direction that part of the field of population genetics is moving in, as the field realizes how limiting and unrealistic analytical models and the coalescent can be (even while they are obviously fast and powerful, and certainly have their uses). We show how to do such forward simulations for bacteria, for which no efficient and flexible simulator exists in 2020, and demonstrate the power of our method by showing a simple spatial model with environmental pressure and ongoing selection that could never be run with the coalescent, and that could obviously be used or extended to address all sorts of interesting questions. The other reasons advanced by the recommender to reject our paper are the following :
(i) A critic that the novel experiment is not reproducible, yet we included the new model in our Github repository associated with this paper at the time of submitting the review; to fully clarify this we now have added a more explicit sentence in the figure caption itself, and in the methods section.
(ii) The recommender was confused about the presence of negative values on a plot with positively defined data. We understood that the recommender was concerned that showing the mean minus std was confusing for the readers because it has negative values, while the empirical values are all positives, so we removed this side of the theoretical expectation (the theoretical line only), as asked. However, we did not understand from the comment that there was an incomprehension: the mean minus standard deviation of a distribution can be negative even if all values are positives. Although not fully informative about a distribution, showing mean +/- std is a common usage. We want to stress here that no experimental points were removed from the figures.
(iii) A missing supplementary figure and another supplementary figure that we unfortunately forgot to update.
(iv) Vague comments such as we “did not address many points the reviewers have made” without detailing which points, while in our opinion we had addressed all the points, or “the figure legends have not improved”, although we did change the figures and figure captions to include the first round of comments.
(v) Surprisingly, the recommender also questions the meaning of a term in a new supplementary figure. This term is not correctly reported by the recommender (”burn-in through capitation” instead of “burn-in through recapitation”), and is a term that appears (with its derivatives) no less than 12 times in the main text, including one section title (”Recapitating and adding neutral mutation”). The concept behind this term is explained in detail in different sections of the paper, including the above-mentioned dedicated section. We do not believe these are fair and constructive critics that could justify a rejection. We did not identify in the first (and only) round of reviews a single comment that would question the scientific quality of our work, nor was the request of solving a biological problem a mandatory one. Most of the reviewers’ comments were based on incomprehension or technical questions, and we believe we have addressed those carefully. For all of these reasons, we are appealing the decision of the recommender and of the managing board to reject bluntly our revised manuscript. Please find attached, the updated version of the manuscript (after the recommender’s comments), which is now online on bioRxiv (version 3).
Sincerely,
Jean Cury and co-authors
Decision by Frederic Bertels, posted 08 Dec 2020
Dear authors,
Thank you for submitting the revised version of the manuscript. Although, I feel like the current version is an improvement, I cannot recommend it for the following reasons:
1. In your reply you wrote “The adaptation of SLiM presented here is not aimed at competing with FastSimBac or ms on “simple” scenarios but rather to open new simulation possibilities”. Indeed this was not clear in the last version you submitted. One of the main reasons this was not clear is that you failed to present data to support the point that SLIM opens up new simulation possibilities. It is indeed important to compare the SLIM code to existing methods but what needs to follow is a detailed analysis of a novel use case or a novel class of use cases that are impossible or difficult to simulate with existing methods or a use case that simply solves an interesting biological problem. The use case you present now, seems to be an interesting one. Yet, currently there is only a brief mention and a figure of the results. There is no analysis and no code to replicate the figure. The reviewers and I feel like the value of the paper really hangs on the detailed analysis of such an example. Without it, the additional value the manuscript provides over existing SLIM manuscripts and existing simulation methods is small. In my opinion this is also the best way to achieve the aim you state at the end of the discussion “We hope that our work here will stimulate a wave of development of simulation-based models for bacterial population genetics.”. Scientists will certainly be animated by an amazing analysis of a simulated evolution experiment that solves an actual biological question. Like it has been done with other scripting languages such as Avida.
2. Unfortunately you have not replied to many points the reviewers have made. For example, one of my comments has been left unanswered. Why are there negative values when you normalize the results? I can see that they have now disappeared, but what happened? Also, Supplementary Figure 11 still has those negative values. Is this intended?
3. Supplementary Figures are not numbered correctly and some Supplementary Figures in the text do not seem to exist (or maybe they exist but have a different number).
4. Supplementary Figure 8 is impossible to understand. Why does A start with 2? What does burn-in through capitation mean? What is shown in the figure (e.g. what are the different colored circles?)?
5. Generally, the figure legends have not improved unfortunately. I wish I could be more positive, but in its current state I cannot recommend the submitted manuscript.
Evaluation round #1
DOI or URL of the preprint: 10.1101/2020.09.28.316869
Version of the preprint: 1
Author's Reply, 30 Nov 2020
Decision by Frederic Bertels, posted 06 Nov 2020
The reviewers find the approach presented here interesting, but criticize that you have not established the specific advantage of the presented approaches over existing approaches. We feel it is important to analyse common use cases where existing approaches fail or cannot be applied. So far the only comment on the advantage of SLiM over the other methods seems to be that SLiM can take circular genomes into account (How much does this matter?). Furthermore, we find it difficult to interpret the data and figures presented in the results section. For example, the data presented in Figure 2: 1. There is no 1 to 1 comparison between the WF expectation and the simulation results. For example, a simulation without recombination would be useful to show that in ideal circumstances the simulations perform as expected. 2. Why/how can the normalization lead to negative values? A better explanation of how the normalization works would be helpful interpreting the figure. It is also unclear what exactly the figures are intended to show. If the main aim of the figure is to show that rescaling does not have an effect on the data, then the figure should show a direct comparison between different scaling factors. Once it is established that the scaling factors do not change the results, SLiM could then be compared to existing methods. In general, as has been pointed out by the reviewers, improved figure legends would help with understanding the presented data. Finally, jargon and abbreviations are used to an extent that the paper becomes difficult to read. In conclusion the manuscript requires very substantial revision in order to be recommended. Importantly, we feel a revision should include data regarding the advantage of SLiM over existing methods.
Additional requirements of the managing board:
As indicated in the 'How does it work?’ section and in the code of conduct, please make sure that:
-Data are available to readers, either in the text or through an open data repository such as Zenodo (free), Dryad or some other institutional repository. Data must be reusable, thus metadata or accompanying text must carefully describe the data.
-Details on quantitative analyses (e.g., data treatment and statistical scripts in R, bioinformatic pipeline scripts, etc.) and details concerning simulations (scripts, codes) are available to readers in the text, as appendices, or through an open data repository, such as Zenodo, Dryad or some other institutional repository. The scripts or codes must be carefully described so that they can be reused.
-Details on experimental procedures are available to readers in the text or as appendices.
-Authors have no financial conflict of interest relating to the article. The article must contain a "Conflict of interest disclosure" paragraph before the reference section containing this sentence: "The authors of this preprint declare that they have no financial conflict of interest with the content of this article." If appropriate, this disclosure may be completed by a sentence indicating that some of the authors are PCI recommenders: “XXX is one of the PCI XXX recommenders.”
Reviewed by anonymous reviewer 1, 06 Nov 2020
Reviewing the paper: Simulations of bacteria populations with SliM
Dear authors, I appreciate your effort in writing this manuscript. Overall I found the manuscript interesting.
Introduction |- Comments
I found the title of the paper a bit deceiving. From the title, I was expecting to see an example of bacterial populations under complex demographic scenarios and selection forces. This paper is more technical. In the first two paragraphs, you explain why simulations are so crucial in bacterial population genomics. Simulation can reveal the past and forecast the new demographic and evolutionary changes of bacterial populations. In your paper, though you do not show any direct evidence of SLiM doing that. You do not show any example where SLiM quantifies the eco-evo dynamics of bacterial populations. In the third paragraph, you compared SLiM to other simulators (e.g., a forward genetic simulator that can simulate complex scenarios including demographics and selection forces, has its language Eidos which makes easily adjustable to simulate bacterial populations).
Methods |-Comments The methods section confused me so much. For this, I'll go step by step. SLim comes together with the following characteristics: 1. Forward simulator 2. It has its own coding language, Eidos, which makes it adjustable for simulating bacterial populations 3. It allows you to simulate bacterial simulation under the assumptions of a Wright-Fisher model and a non-WF framework. This is quite clear to me. Comment: In this manuscript, you are performing simulations of bacterial populations under the non-WF framework, but you do not validate the non-WF results with any experimental data.
Methods
|-Horizontal gene transfer, recombination and circularity - Comments
Horizontal gene transfer: The exchange of pieces of DNA between different organisms. The piece can be inserted at a random site or a specific site. If the incoming fragment is homologous, then the piece can be incorporated in a way that is similar to gene conversion to eukaryotes, where you do not have a reciprocal exchange of genetic material.
Comment: I see the importance of taking into consideration gene conversion, but you can potentially cite a paper reflecting its importance in the adaptation of bacterial populations, together with the frequency of gene conversion and homologous recombination. Also, you talk so much about gene conversion which at the end you do not consider it in your results, except if you refer to recombination as gene conversion which I doubt. This isn't very clear. You rightly claim that SLiM is superior to other programs because it can simulate gene conversion and because you consider the bacterial chromosome is circular. Why is this important? It is known that a bacterial chromosome, in any case, looks like a smear, a chaotic construction where DNA helixes are entangled with each other. Also, later in the paper, you counter-attack your argument of gene conversion by writing. Because we simulate the entire population; it is not possible to use gene conversion at a significant rate, otherwise ms crashes, thus there is no recombination in "burn-in"
Methods |-Burn-in - Comments
It is desirable to start a simulation with a population that is in a mutation-drift equilibrium. We have a mutation-drift equilibrium when both the mutation rate and the effective population size are stable. In a mutation drift equilibrium, the rate that the variation is lost due to drift is the same that is gained due to mutation.
Comments: I do not understand what does it mean when you say that the population size is larger than the time-span of interest guess you mean the effective population size that is needed to reach a mutation drift equilibrium is very high. Could you clear this out?
Methods |- Simulation rescaling - Comments Here you discuss the effect of rescaling into the summary statistics of the program. It's quite clear to me.
Methods |- Simulation protocol - Comments
Overall the simulation protocol is detailed and well explained. Many times, however, I was getting errors when I tried to copy-paste the code in the SLiMgui (e.g., ERROR (EidosSymbolTable::_GetValue): undefined identifier genomeSize. This error has invalidated the simulation; it cannot be run further. Once the script is fixed, you can recycle the simulation and try again) I suggest making the code more accessible, so when we test the code of the paper not to paste the line numbers as well. However, I see the importance of enumeration. In the end, I used your GitHub code where enumeration is hard to be followed.
Results The Results are quite straight forward. However, when I was reading your introduction, I was prepared for a different type of results. You did what you wrote about at the end of the introduction (you introduced the model, and that model behaves according to WF-model). Still, you also present a non-WF model whose results you do not validate from experimental data.
Figure1: rescaling ~ CPU time and memory Figure2: SFS ~ rescaling Figure3: LD ~ rescaling Figure 4: recombination rater & tract length ~ CPU & memory Figure 5: SFS ~ recombination rate Figure 6:
Comment: With the caption of your figures, you should convey the main result of the figure to be easier for the reader to skim through your soon to be published. For example, in Figure 1, you could write that by increasing the rescaling factor you observer faster CPU time, and less memory and that nonWF pops are being faster.
Discussion
In the discussion, you summarise your results and refer to the drawbacks of your simulator. I could not even find a typo. In general, I have to admit that I admire your efforts. The paper is neat, well structured, even the bibliography is written accurately. However, there is a space for improvement. Your methods section I believe that needs to be written more clearly. There are several points where the reader gets confused. You have to make from the introduction very clear your points, do not refer to gene conversion as your strong point since it is not, clear out what do you mean by recombination, pass out that this is technical paper.
https://doi.org/10.24072/pci.evolbiol.100356.rev11Reviewed by anonymous reviewer 2, 28 Oct 2020
Reviewed by anonymous reviewer 3, 22 Oct 2020
This manuscript describes how to adapt the popular simulator SLiM to bacteria, especially to the bacterial mode of recombination. I had wondered about this possibility myself in the past, and I am delighted to see this preprint and the described protocol. However, I see several possibilities for improving the manuscript to better highlight the improvements of the described approach compared to existing approaches.
The manuscript would greatly profit from an overview figure that explains the underlying model and the different parameters used and how they go into the simulation.
The main advantage of the described approach should be presented with an example and discussed. So far, only simulations with comparisons to other programs are done, and they show convincingly that the SLiM approach works well. However, it is not obvious which advantages the presented approach has compared to ms and FastSimBac. Maybe one more complex simulation that includes selection or population structure could be added in the end to show an application of the approach. The advantages over previous approaches could also be added to the "Discussion" section.
Section 2.2.1 "mean recombination tract length of 10kb" First, the distribution could be mentioned here already, although this can be seen in the code. My main point is, however, that this value appears quite large. E.g., unselected recombination events found in 10.1371/journal.ppat.1002745 are on average 2kbp, most of the recombinations inferred in 10.1128/mBio.02494-18 are below 10kb, and the average length of homologous recombination fragments inferred in E. coli is ~500bp (10.1186/1471-2164-13-256). The simulation is presented for parameters from S. agalactiae where the mean length is even above 100kb, and this paper is based on selected recombination events, whereas unselected events should provide the parameters for the simulation (see 10.1371/journal.ppat.1002745 for the difference). Although, I understand that these parameters can be adjusted, I wondered how the simulations perform for shorter length.
Section 2.2.1 It is not clear to me how the source individual for the recombination event is chosen. Since offspring is directly added to the population, is it possible, that generated recombinants can already be the source individual for recombinants generated later in the same generation?
The authors should mention the recently released simulator CoreSimul (10.1186/s12859-020-03619-x), maybe in the introduction. If feasible, it would be interesting to see how it compares to SLiM.
Additional comments: Section 2.1.2 "Because we simulate the entire population, it is not possible to use gene conversion at a significant rate, otherwise ms crashes, thus there is no recombination in burn-in." Maybe you can be more precise and describe why ms crashed, would more RAM solve the issue? Which population size would the feasible with ms? Section 2.1.3 "The rescaling factor must also be applied to the duration of the simulation (and the duration of different events that might occur), so that the effects of drift remains similar." Maybe it could be described explicitly how the length and events should be increased or decreased. Section 2.2.1 "constant 11" Should it read "constant 1"?
https://doi.org/10.24072/pci.evolbiol.100356.rev13



