
Shedding light on genomic divergence along the speciation continuum
Drivers of genomic landscapes of differentiation across Populus divergence gradient
Abstract
Recommendation: posted 25 January 2023, validated 25 January 2023
Llaurens, V. (2023) Shedding light on genomic divergence along the speciation continuum. Peer Community in Evolutionary Biology, 100488. https://doi.org/10.24072/pci.evolbiol.100488
Recommendation
The article “Drivers of genomic landscapes of differentiation across Populus divergence gradient” by Shang et al. describes an amazing dataset where genomic variations among 21 pairs of diverging poplar species are compared. Such comparisons are still quite rare and are needed to shed light on the processes shaping genomic divergence along the speciation gradient. Relying on two hundred whole-genome resequenced samples from 8 species that diverged from 1.3 to 4.8 million years ago, the authors aim at identifying the key factors involved in the genomic differentiation between species. They carried out a wide range of robust statistical tests aiming at characterizing the genomic differentiation along the genome of these species pairs. They highlight in particular the role of linked selection and gene flow in shaping the divergence along the genomes of species pairs. They also confirm the significance of introgression among species with a net divergence larger than the upper boundaries of the grey zone of speciation previously documented in animals (da from 0.005 to 0.02, Roux et al. 2016). Because these findings pave the way to research about the genomic mechanisms associated with speciation in species with allopatric and parapatric distributions, I warmingly recommend this article.
References
Roux C, Fraïsse C, Romiguier J, Anciaux Y, Galtier N, Bierne N (2016) Shedding Light on the Grey Zone of Speciation along a Continuum of Genomic Divergence. PLOS Biology, 14, e2000234. https://doi.org/10.1371/journal.pbio.2000234
Shang H, Rendón-Anaya M, Paun O, Field DL, Hess J, Vogl C, Liu J, Ingvarsson PK, Lexer C, Leroy T (2023) Drivers of genomic landscapes of differentiation across Populus divergence gradient. bioRxiv, 2021.08.26.457771, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.08.26.457771

The recommender in charge of the evaluation of the article and the reviewers declared that they have no conflict of interest (as defined in the code of conduct of PCI) with the authors or with the content of the article. The authors declared that they comply with the PCI rule of having no financial conflicts of interest in relation to the content of the article.
This work was supported by a fellowship from the China Scholarship Council (CSC) to Huiying Shang, Swiss National Science Foundation (SNF) grant no.31003A_149306 to Christian Lexer, doctoral programme grant W1225-B20 to a faculty team including Christian Lexer, and the University of Vienna.
Evaluation round #2
DOI or URL of the preprint: https://doi.org/10.1101/2021.08.26.457771
Version of the preprint: 4
Author's Reply, 12 Jan 2023
Decision by Violaine Llaurens , posted 13 Oct 2022, validated 14 Oct 2022
, posted 13 Oct 2022, validated 14 Oct 2022
Dear authors,
First, I would like to apologize for the delay in the peer-review process. The three reviewers already solicited for the previous version of the manuscript have now all evaluated the changes made to your manuscript. All three reviewers and myself acknowledge the substantial effort made to shorten and clarify the manuscript. Most previously-raised issues have been successfully addressed. Nevertheless, some reviewers still have reservations and their comments need to be addressed. In particular, I agree with one out of the three reviewers about the section on the comparison of the landscapes and its putative conservation: this section is still a bit inconclusive and the conservation of the landscape across the comparison is not straightforward from the results shown in the manuscript. The effect of linked selection vs. background selection on these landscapes is also potentially an original result but is not explicit enough. Pending you successfully address these last comments, I would be able to recommend your manuscript for PCI evolutionary biology.
Sincerely yours,
Violaine Llaurens
Reviewed by Camille Roux, 08 Oct 2022
Reviewed by Steven van Belleghem, 05 Aug 2022
The authors fully resolved my earlier comments and did an amazing job at improving the structure and presentation of their manuscript. I think their result are of value to a broad audience interested in adaptation, divergence and population genomics. I only have a few small further remarks.
Box 1 is a very helpful way of presenting the expected patterns! I think, perhaps, it would be helpful to clarify that the identification of the patterns may be dependent on the (early) timing of the sampling relative to the time of divergence. For example, I would expect the increased Fst relative to background in scenario 2 to show up only in a certain time window of divergence. If populations have been separated too long, this signal would get lost. In scenario 3, I believe Dxy should only be reduced if selection happened before the divergence event.
Figure 2b is a brilliant addition.
As before, it is great to see the presentation of how the relationship between these statistics changes along the divergence gradient in Figure 4 and how these changes in relationships match with theoretical predictions.
Supplementary Note 3 regarding the fd analysis does not seem to be referenced or mentioned in the main text.
https://doi.org/10.24072/pci.evolbiol.100488.rev22Reviewed by anonymous reviewer 1, 26 Sep 2022
In this manuscript, Shang and colleague study a radiation of Populus trees, to decipher the most likely scenario explaining speciation in this group. Specifically, by sequencing individuals from 7 species, they test 4 different speciation scenarios (divergence with gene flow, allopatric speciation, recurrent selection and balancing selection). They conclude that, in Populus, allopatric speciation fits most of the genetic differentiation observed in the genome and is the most likely mode of speciation in the genus. Authors also detected two introgression events in the radiation.
The study system is interesting and the authors use state of the art methodologies to answer their questions.
The authors seem to have considered reviewers comments on a previous draft but it is difficult to tell given that they did not provide a response letter of any kind.
It seems that the manuscript has been extensively rewritten and is now clearer. I however still have a couple of concerns with the current version of the manuscript.
First, the distinction between model 3 and 4 involves more quantitative testing than qualitative testing, as the signs of the correlation between statistics are similar but just of a different magnitude. I therefore did not understand how the authors manage to distinguish between these two scenarios looking strictly at genome wide correlations?
Second, the authors use the term 'genomic landscapes' to describe the variation of many statistics along the genome. I find it a source of confusion personally.
Third, the authors recurrently mention that these landscapes are heterogeneous, but never illustrate it or test it. I think a test could be done to verify if the windows have higher, lower, or similar levels of divergence (or any other statistics) than the rest of the genome and see if it a different distribution than what can be expected by chance (using simulation data with an allopatric model possibly). I feel it is quite important given the aim of the paper is to understand why there might be heterogeneity in these landscapes.
Fourth, the authors mention repeatedly that these genomic landscapes are conserved across the speciation continuum based on correlations of statistics across the genome. However, the correlation coefficients that the authors find are significant but low for most statistics, implying that these landscape are somewhat similar but also somewhat different. I think the authors could do a better job at quantifying the amount of overlap across species. Also, some correlation coefficients decrease with time, indicating that they are actually not conserved.
Finally, I do not grasp the novelty of the current study. The message I get is 'Heterogeneous landscapes emerge because of different evolutionary process'. I feel that a deeper comparison of the authors' results with past literature in the Discussion section would clarify this point.
I personally felt that the manuscript lacks clarity on the points I mentioned above and therefore do not recommand publication of the manuscript in its current form.
Specific comments:
lines 54-55: Too vague. Strictly speaking we understand this now: Mutation, selection, gene flow and drift.
line 62: What are 'hotspots' of elevated genetic differentiation?
lines 72-74: what does 'highly heterogeneous genomic landscapes' mean?
lines 225-227: What else does shape it then?
line 240: I would argue that the genomic patterns are 'somewhat' conserved here.
https://doi.org/10.24072/pci.evolbiol.100488.rev23Evaluation round #1
DOI or URL of the preprint: https://doi.org/10.1101/2021.08.26.457771
Version of the preprint: 2
Author's Reply, 19 Jul 2022
Decision by Violaine Llaurens, posted 17 Nov 2021
Dear authors,
I found that your manuscript the genomic landscapes of differentiation across Populus speciation continuum brings original and news insights on the genomic processes involved in population differentiation. The quality of the datasets and of the analyses carried out have impressed the three reviewers and myself. Nevertheless, the manuscript is quite long and is sometimes difficult to read. Importantly the take-home messages are not straightforward to understand, preventing my recommendation at this point.
The three reviewers have listed specific points that should be addressed, and I would be happy to evaluate again an improved version of the manuscript, clarifying the results and the main conclusions drawn from your study. The number of analyses and figures reported is very large, and you may consider sorting out the most important ones, and move the less important ones in the supplementary section.
Overall, you may especially aim at reaching a larger audience of evolutionary biologists interested in speciation, including readers lacking background in population genomics.
Sincerely yours,
Violaine Llaurens
Specific comments:
Line 33 : You may briefly define what you mean here by ‘genomic landscape’.
Line 44 and 47: Again providing a hint about what ‘linked selection ‘ and ‘background selection’ mean here, and how these selective forces might influence genomic divergence between species seems necessary.
Line 48: It is unclear to me how you can infer that the divergent sorting happened before speciation ?
Line 49-51: this conclusion is very vague and a bit circular (studying the genomic patterns informs on genomic landscape of differentiation?).
Line 61-63: this sentence is too vague, what can of ‘overall drivers’ are you referring to?
Line 66: ‘local adaptation AND/OR reproductive isolation
Line 70: i.e. should be italics
Line 83: It is unclear what did you mean by ‘genomic characteristic influenced by life history traits’ ? Would selfing rate be an example of such life history traits? Because selfing in plant would decrease heterozygosity throughout the genomes? It is worth being more explicit here in my opinion.
The processes listed line 82-83 are nevertheless not independent from adaptive processes: for instance, variation of recombination can be linked to adaptation. Maybe the beginning of this sub-section should be rephrased.
Lines 125-139 could be moved to a box containing Fig. 1
Line 151: add parenthesis in the reference Burri (2017a)
Line 155: remove ‘This is due to the fact that’
Line 147-162: could also be grouped together in a single Box summarizing the expectations and Fig. 1.
Line 182: ‘open questions’ it would help the reader to focus on the main question addressed in the manuscript to ease the understanding, rather than providing such highly general statements.
Line 170-182: Overall this section should be moved to the Material and method section, this would make the introduction shorter and more impactful.
Lines 201-208 : This could move to the material and method section as well.
Lines 213-221: this seems a bit descriptive and could be removed. A brief mention on fig. S4 separating the most recently diverged species would be sufficient.
Line 252: ‘in A previous study (Shang et al. 2020)’
Line 287-289: replace ‘significantly’ by ‘significant’ (line 287 and line 289) and ‘evident’ by ‘obvious’
Line 291: ‘indicates THAT gene density is…’
Line 302: ‘To have knowledge of…’ it is important to avoid vague sentences, and to guide readers into the different questions addressed in the sub-sections of the manuscript.
Line 305: It is unclear why you used only 5 species while you have data on many other ones? Is it simply for showing a simplified figure? I guess that using the whole datasets would even strengthen the point you are making on the conserved pattern of differentiation?
You may consider reducing the number of figures in the main document, there is a large amount of data presented, I guess it would improve the manuscript by emphasizing more some results and reducing the number of analyses presented. Some figures could be moved in the supplementary material, to help reader focus on the main results. Maybe figure 5 is slightly redundant with fig.4, and could be moved to supplementary for instance?
A mentioned by one of the reviewers, the three last sections of the manuscript are quite difficult to understand and are probably the most important of the manuscript. It is hard for the reader to get the exact information gained by the different analyses, how much they are redundant or provide independent evidences of the same phenomenon.
Line 414-416: this conclusion is surprising: while pointing out the limit of your analyses is necessary, highlighting that the scenario tested are naïve prevents the reader to get the general message of your study.
Line 424: you may remove the emphasis; the dataset is impressive, there is no need claiming superiority. Replace by ‘we provided a relevant and ambitious case-study’.
Line 426: support THE role of linked selection
Line 430 : ‘play a role’ is vague and participate to the lack of clarity on the main findings of the study.
Line 433-436: I am not sure this sentence is useful, it also provides a bit circular argument.
Reviewed by Camille Roux, 12 Nov 2021
Reviewed by Steven van Belleghem, 03 Oct 2021
Huiying Shang and colleagues present a detailed study on the genomic divergence landscapes between pairs of Populus tree species. They use more than two hundred whole-genome resequenced samples from 8 species to study genetic divergence along a continuum of speciation. By correlating measures of nucleotide diversity, relative divergence, absolute divergence, and recombination rate they set out a number of hypothesis that each well define how these relationships are expected to change as divergence increases between species. Doing this, they identify the common drivers of heterogeneity in genomic divergence to be background selection, with contributions of ancestral polymorphisms, gene flow and potentially selection.
I think the authors did an excellent job at introducing these complex concepts and outlining their hypothesis that can be neatly dissected with their data. The number of possible expected relationships between population genomic statistics is high and I was truly impressed how well the authors did at explaining their expectations and being able to follow how their results fit to them. I think the analysis and results are solid, but please allow me to give a few remarks that I hope could further increase the accessibility of this manuscript to a broad public.
First, I very much like Figure 1. Panel A shows expected correlations within or between species, whereas panel B shows how these relationships may change over time. In the results, however, results relating to panel A are presented at the end and it would have been more logical to me to have presented those results first (regarding that section, I would also suggest including the figures in the main text, rather than the supplement, because a quantification of each hypothesis is really unique! I think doing this could also improve understanding how much each of the different factors contributed to the divergence landscape.). Next, In Fig4 and Fig6 I would suggest improving the link with figure 1, by showing the results in the same order as in Fig1 and clearly pointing the reader to how they match Fig1 and what it tells about the drivers of genomic divergence.
Second, I think there’s a missed opportunity to better quantify the extent of gene flow between species, compared to using IBD and TreeMix. Would it be possible to calculate ABBA-BABA type of statistics (e.g. fd https://github.com/simonhmartin/genomics_general/blob/master/ABBABABAwindows.py) to understand what fraction of the differentiation heterogeneity results from admixture? Another set of analyses could include running SweepFinder in each population to identify common and unique selective sweeps.
Third, do reads of more distant species map equally well to the reference genome. It might be good to verify this, so that no correlation biases are created because more distant species have less reads mapping, resulting into reduced diversity values.
Fourth, regarding the title “Conserved genomic landscapes of differentiation”, the point of conservation is a bit lost to me (after the abstract) as most of the later figures visualize how relationships between statistics change as species diverge. I might thus suggest presenting a more direct quantification of the conservation of the differentiation landscape. I do agree that the drivers of genomic differentiation may be highly conserved.
Small comments:
Be careful in the use of ‘linked selection’ and ‘background selection’. They are not the same and I think they are sometimes used as interchangeable (e.g. in the abstract L44 and L46).
L87-92: Also observed in Heliconius (and including introgression) (Martin et al. 2019 Plos Biology; Edelman et al. 2019 Science; Van Belleghem et al. 2021 Evolution)
L124: “different scenarios can be hypothesized regarding the extent of local gene flow…”. I found this sentence confusing because the four scenarios do not just focus on gene flow, but include balancing selection, background selection and selective sweeps. This becomes clear further in the manuscript, but I would argue to modify the sentence here.
L145: “…polygenetic adaptation from standing genetic variation”. Does this include patterns arising from linked selection in the common ancestor?
L153: “recombination (r)”
L154: I would suggest “across the speciation continuum”
L154: “…negative correlation of Fst and r become increased with advancing differentiation…”. Is this assuming no gene flow? I imagine that with gene flow, this correlation can also become increased.
L158: “The positive correlations between p and r should remain highly correlated with each other under background selection”. Can selective sweeps also contribute here to that relationship?
L178: Populus trichocarpa
L179: P. trichocarpa
L226: Fig. 2c instead of Fig. 1c?
L229: Can shared haplotypes in the IBD analysis also result from incomplete lineage sorting?
Fig3a: Why do introgression edges go from tree edges to tree tips and not tree edge to tree edge? (Maybe I am just not familiar with the method)
Fig4e-f: Would it be possible to visualize and add the genome-wide correlation of p and r?
L285: Regarding the low p but high r in P. grandidentata, I noticed that this species has undergone consistent population decline. Could that have affected p independently from r?
L318: “correlation coefficients for…”. Correlation with what in this case? I assume with divergence level, but this took me some time to understand.
L341: Is “respectively” needed here?
L342: Again, I would suggest “across the speciation continuum”
L352: “…expect negative correlations between Fst and p or r and the relationships become stronger as da increases”. Would there not be a limit to this, e.g., when Fst reaches 1?
L353-354: This is a difficult sentence to me. Should it read: “Besides, the positive correlations between Dxy and p should be highly correlated with da …”?
L359-361: “…p and Dxy showed significantly positive correlations while the trend became weaker as divergence increased”. Is this not expected as the proportional contribution of ancestral p to Dxy and the current p will decrease over time? Hence, the correlation will decrease?
Fig6: I would suggest fixing the y-axis so that all plots are readily comparable.
L387: There seems something wrong with this sentence.
L405-406: It is unclear to me how support was found for reproductive barriers.
https://doi.org/10.24072/pci.evolbiol.100488.rev12
Reviewed by anonymous reviewer 1, 10 Nov 2021
In this manuscript, Shang and colleague study a radiation of Populus trees, to decipher the most likely scenario explaining speciation in this group. Specifically, by sequencing individuals from 7 species, they test 4 different speciation scenarios (divergence with gene flow, allopatric speciation, background selection and balancing selection). They conclude that, in Populus, allopatric speciation fits most of genetic differentiation observed in the genome and is the most likely mode of speciation in the genus. Authors also detected two introgression events in the radiation.
The study system is interesting and the authors use state of the art methodologies to answer their questions.
However, the authors explain very succinctly the two last models they test for and the associations of statistics they expect to emerge if these models are true. Specifically I did not understand model 4 (balancing selection) and how this model differed from model 1. Also, the distinction between model 3 and 4 involves more a quantitative testing than a qualitative testing, as the signs of the correlations between genetic statistics are similar but just of a different magnitude. I therefore did not understand how the author did manage to distinguish these two scenarios?
I personally felt that the manuscript is poorly written in parts, making some sections unintelligible. Indeed, I did not understand a couple of result sections (Conserved genomic landscapes across the continuum of divergence, Correlated patterns of genome-wide variation across speciation continuum, and Scenarios of genomic patterns of differentiation) because of both the writing, and the many reasoning shortcuts used in these sections.
I therefore do not recommand publication of the manuscript in its current form.
I think the above mentioned sections of the manuscript need to be rewritten. This is more of a personal preference, but the flow of the manuscript could be changed to present the main results of the manuscript (i.e. the hypothesis testing) earlier on. In that line of thinking I would put the Between species variability in demographic trajectories section in the SI, and bring from the SI Figure S11 which I feel is central to the results of this paper.
Specific comments:
lines 143-145: expand reasoning.
line 148: why is this expected?
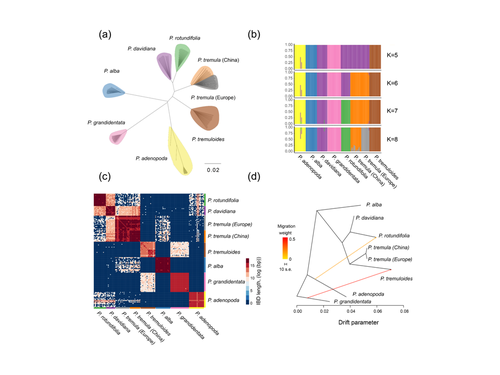
Figure 2c: There is more colour than cluster number for K=5. Why is there green colour in pade and pdav individuals?
line 287: gene density appears from nowhere. Integrate it in the introduction.
lines 289-291: Why? expand reasoning.
lines 305-306: How where selected the 'representative' species pair?
lines 316-317: reformulate. Fst along the genome. What does 'independant' refers to for species pairs?
lines 317-318: I do not understand this sentence.
lines 318-322: I do not understand this sentence.
lines 328-330: expend reasoning.
line 342: the speciation continuum?
lines 345-346: I don't understand this sentence.
https://doi.org/10.24072/pci.evolbiol.100488.rev13



