
PYHÄJÄRVI Tanja
- Department of Forest Sciences, University of Helsinki, Helsinki, Finland
- Adaptation, Evolutionary Applications, Evolutionary Theory, Expression Studies, Genetic conflicts, Genome Evolution, Genotype-Phenotype, Hybridization / Introgression, Molecular Evolution, Population Genetics / Genomics
- recommender
Recommendation: 1
Reviews: 2
Recommendation: 1

Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network
Early and late flowering gene expression patterns in maize
Recommended by Tanja Pyhäjärvi based on reviews by Laura Shannon ? and 2 anonymous reviewersArtificial selection experiments are key experiments in evolutionary biology. The demonstration that application of selective pressure across multiple generations results in heritable phenotypic changes is a tangible and reproducible proof of the evolution by natural selection.
Artificial selection experiments are used to evaluate the joint effects of selection on multiple traits, their genetic covariances and differences in responses in different environments. Most studies on artificial selection experiments report and base their analyses on phenotypic changes [1]. More recently, changes in allele frequency and other patterns of molecular genetic diversity have been used to identify genomic locations where selection has had an effect. However, so far the changes in gene expression have not been in the focus of artificial selection experiment studies (see [2] for an example though).
In plants, one of the most famous artificial selection experiments is the Illinois Corn Experiment where maize (Zea mays) is selected for oil and protein content [3], but in addition, similar experiments have been conducted also for other traits in maize. In Saclay divergent selection experiment [4] two maize inbred lines (F252 and MBS847) have been selected for early and late flowering for 13 generations, resulting in two week difference in flowering time.
In ”Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network ” [5] Maud Tenaillon and her coworkers study the gene expression differences among these two independently selected maize populations. Their experiments cover two years in field conditions and they use samples of shoot apical meristem at three different developmental stages: vegetative, transitioning and reproductive. They use RNA-seq transcriptome level differences and qRT-PCR for gene expression pattern investigation. The work is continuation to earlier genetic and phenotypic studies on the same material [4, 6].
The reviewers and I agree that dataset is unique and its major benefit is that it has been obtained from field conditions similar to those that species may face under natural setting during selection. Their tissue sampling is supported by flowering time phenotypic observations and covers the developmental transition stage, making a good effort to identify key transcriptional and phenotypic changes and their timing affected by selection.
Tenaillon et al. [5] identify more than 2000 genes that are differentially expressed among early and late flowering populations. Expectedly, they are enriched for known flowering time genes. As they point out, differential expression of thousands of genes does not mean that they all were independently affected by selection, but rather that the whole transcriptional network has shifted, possibly due to just few upstream or hub-genes. Also, the year-to-year variation had smaller effect in gene expression compared to developmental stage or genetic background, possibly indicating selection for stability across environmental fluctuation for such an important phenotype as flowering time.
Another noteworthy observation is that they find convergent patterns of transcriptional changes among the two selected lines. 115 genes expression patterns are shifted due to selection in both genetic backgrounds. This convergent pattern can be a result of either selection on standing variation or de novo mutations. The data does not allow testing which process is underlying the observed convergence. However, their results show that this is an interesting future question that can be addressed using genotype and gene expression data from the same ancestral and derived material and possibly their hybrids.
References
[1] Hill, W. G., & Caballero, A. (1992). Artificial selection experiments. Annual Review of Ecology and Systematics, 23(1), 287-310. doi: 10.1146/annurev.es.23.110192.001443
[2] Konczal, M., Babik, W., Radwan, J., Sadowska, E. T., & Koteja, P. (2015). Initial molecular-level response to artificial selection for increased aerobic metabolism occurs primarily through changes in gene expression. Molecular biology and evolution, 32(6), 1461-1473. doi: 10.1093/molbev/msv038
[3] Moose, S. P., Dudley, J. W., & Rocheford, T. R. (2004). Maize selection passes the century mark: a unique resource for 21st century genomics. Trends in plant science, 9(7), 358-364. doi: 10.1016/j.tplants.2004.05.005
[4] Durand, E., Tenaillon, M. I., Ridel, C., Coubriche, D., Jamin, P., Jouanne, S., Ressayre, A., Charcosset, A. and Dillmann, C. (2010). Standing variation and new mutations both contribute to a fast response to selection for flowering time in maize inbreds. BMC evolutionary biology, 10(1), 2. doi: 10.1186/1471-2148-10-2
[5] Tenaillon, M. I., Seddiki, K., Mollion, M., Le Guilloux, M., Marchadier, E., Ressayre, A. and Dillmann C. (2019). Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network. BioRxiv, 461947 ver. 5 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/461947
[6] Durand, E., Tenaillon, M. I., Raffoux, X., Thépot, S., Falque, M., Jamin, P., Bourgais A., Ressayre, A. and Dillmann, C. (2015). Dearth of polymorphism associated with a sustained response to selection for flowering time in maize. BMC evolutionary biology, 15(1), 103. doi: 10.1186/s12862-015-0382-5
Reviews: 2
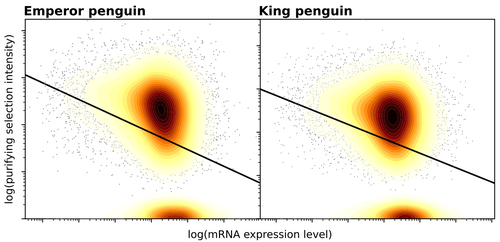
Gene expression is the main driver of purifying selection in large penguin populations
Purifying selection on highly expressed genes in Penguins
Recommended by Bruce Rannala based on reviews by Tanja Pyhäjärvi and 1 anonymous reviewerGiven the general importance of protein expression levels, in cells it is widely accepted that gene expression levels are often a target of natural selection and that most mutations affecting gene expression levels are therefore likely to be deleterious [1]. However, it is perhaps less obvious that the strength of selection on the regulated genes themselves may be influenced by their expression levels. This might be due to harmful effects of misfolded proteins, for example, when higher protein concentrations exist in cells [2]. Recent studies have suggested that highly expressed genes accumulate fewer deleterious mutations; thus a positive relationship appears to exist between gene expression levels and the relative strength of purifying selection [3].
The recommended paper by Trucchi et al. [4] examines the relationship between gene expression, purifying selection and a third variable -- effective population size -- in populations of two species of penguin with different population sizes, the Emperor penguin (Aptenodytes forsteri) and the King penguin (A. patagonicus). Using transcriptomic data and computer simulations modeling selection, they examine patterns of nonsynonymous and synonymous segregating polymorphisms (p) across genes in the two populations, concluding that even in relatively small populations purifying selection has an important effect in eliminating deleterious mutations.
References
1] Gilad Y, Oshlack A, and Rifkin SA. 2006. Natural selection on gene expression. Trends in Genetics 22: 456-461. https://doi.org/10.1016/j.tig.2006.06.002
[2] Yang JR, Liao BY, Zhuang SM, and Zhang J. 2012. Protein misinteraction avoidance causes highly expressed proteins to evolve slowly. Proceedings of the National Academy of Sciences 109: E831-E840. https://doi.org/10.1073/pnas.1117408109
[3] Duret L, and Mouchiroud D (2000). Determinants of substitution rates in mammalian genes: expression pattern affects selection intensity but not mutation rate. Molecular Biology and Evolution 17; 68-070. https://doi.org/10.1093/oxfordjournals.molbev.a026239
[4] Trucchi E, Massa P, Giannelli F, Latrille T, Fernandes FAN, Ancona L, Stenseth NC, Obiol JF, Paris J, Bertorelle G, and Le Bohec, C. 2023. Gene expression is the main driver of purifying selection in large penguin populations. bioRxiv 2023.08.08.552445, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.08.08.552445

Modularity of genes involved in local adaptation to climate despite physical linkage
Differential effect of genes in diverse environments, their role in local adaptation and the interference between genes that are physically linked
Recommended by Sebastian Ernesto Ramos-Onsins based on reviews by Tanja Pyhäjärvi and 1 anonymous reviewerThe genome of eukaryotic species is a complex structure that experience many different interactions within itself and with the surrounding environment. The genetic architecture of a phenotype (that is, the set of genetic elements affecting a trait of the organism) plays a fundamental role in understanding the adaptation process of a species to, for example, different climate environments, or to its interaction with other species. Thus, it is fundamental to study the different aspects of the genetic architecture of the species and its relationship with its surronding environment. Aspects such as modularity (the number of genetic units and the degree to which each unit is affecting a trait of the organism), pleiotropy (the number of different effects that a genetic unit can have on an organism) or linkage (the degree of association between the different genetic units) are essential to understand the genetic architecture and to interpret the effects of selection on the genome. Indeed, the knowledge of the different aspects of the genetic architecture could clarify whether genes are affected by multiple aspects of the environment or, on the contrary, are affected by only specific aspects [1,2].
The work performed by Lotterhos et al. [3] sought to understand the genetic architecture of the adaptation to different environments in lodgepole pine (Pinus contorta), considering as candidate SNPs those previously detected as a result of its extreme association patterns to different environmental variables or to extreme population differentiation. This consideration is very important because the study is only relevant if the studied markers are under the effect of selection. Otherwise, the genetic architecture of the adaptation to different environments would be masked by other (neutral) kind of associations that would be difficult to interpret [4,5]. In order to understand the relationship between genetic architecture and adaptation, it is relevant to detect the association networks of the candidate SNPs with climate variables (a way to measure modularity) and if these SNPs (and loci) are affected by single or multiple environments (a way to measure pleiotropy).
The authors used co-association networks, an innovative approach in this field, to analyse the interaction between the environmental information and the genetic polymorphism of each individual. This methodology is more appropriate than other multivariate methods - such as analysis based on principal components - because it is possible to cluster SNPs based on associations with similar environmental variables. In this sense, the co-association networks allowed to both study the genetic and physical linkage between different co-associations modules but also to compare two different models of evolution: a Modular environmental response architecture (specific genes are affected by specific aspects of the environment) or a Universal pleiotropic environmental response architecture (all genes are affected by all aspects of the environment). The representation of different correlations between allelic frequency and environmental factors (named galaxy biplots) are especially informative to understand the effect of the different clusters on specific aspects of the environment (for example, the co-association network ‘Aridity’ shows strong associations with hot/wet versus cold/dry environments).
The analysis performed by Lotterhos et al. [3], although it has some unavoidable limitations (e.g., only extreme candidate SNPs are selected, limiting the results to the stronger effects; the genetic and physical map is incomplete in this species), includes relevant results and also implements new methodologies in the field. To highlight some of them: the preponderance of a Modular environmental response architecture (evolution in separated modules), the detection of physical linkage among SNPs that are co-associated with different aspects of the environment (which was unexpected a priori), the implementation of co-association networks and galaxy biplots to see the effect of modularity and pleiotropy on different aspects of environment. Finally, this work contains remarkable introductory Figures and Tables explaining unambiguously the main concepts [6] included in this study. This work can be treated as a starting point for many other future studies in the field.
References
[1] Hancock AM, Brachi B, Faure N, Horton MW, Jarymowycz LB, Sperone FG, Toomajian C, Roux F & Bergelson J. 2011. Adaptation to climate across the Arabidopsis thaliana genome. Science 334: 83–86. doi: 10.1126/science.1209244
[2] Wagner GP & Zhang J. The pleiotropic structure of the genotypephenotype map: the evolvability of complex organisms. Nature Review Genetics 12: 204–213. doi: 10.1038/nrg2949
[3] Lotterhos KE, Yeaman S, Degner J, Aitken S, Hodgins K. 2018. Modularity of genes involved in local adaptation to climate despite physical linkage. bioRxiv 202481, ver. 4 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/202481
[4] Lotterhos KE & Whitlock MC. 2014. Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Molecular Ecology 23: 2178–2192. doi: 10.1111/mec.12725
[5] Lotterhos KE & Whitlock MC. 2015. The relative power of genome scans to detect local adaptation depends on sampling design and statistical method. Molecular Ecology 24: 1031–1046. doi: 10.1111/mec.13100
[6] Paaby AB & Rockman MV. 2013. The many faces of pleiotropy. Trends in Genetics 29: 66-73. doi: 10.1016/j.tig.2012.10.010