
KISHINO Hirohisa
- Laboratory of Biometry and Bioinformatics, The University of Tokyo, Tokyo, Japan
- Genome Evolution, Molecular Evolution, Morphological Evolution, Phylogenetics / Phylogenomics, Phylogeography & Biogeography, Population Genetics / Genomics
- recommender
Recommendations: 2
Reviews: 0
Recommendations: 2
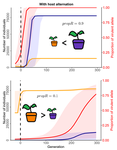
Impact of ploidy and pathogen life cycle on resistance durability
Durability of plant resistance to diploid pathogen
Recommended by Hirohisa Kishino based on reviews by Loup Rimbaud and 1 anonymous reviewerDurability of plant resistance to diploid pathogen Hirohisa Kishino Based on the population genetic and epidemiologic model, Saubin et al. (2021) report that the resistant hosts generated by the breeding based on the gene-for-gene interaction is durable much longer against diploid pathogens than haploid pathogens. The avr allele of pathogen that confers the resistance is genetically recessive. The heterozygotes are not recognized by the resistant hosts and only the avr/avr homozygote is adaptive. As a result, the trajectory of avr allele frequency becomes more stochastic due to genetic drift. Although the paper focuses on the evolution of standing polymorphism, it seems obvious that the adaptive mutations in pathogen have much larger probability of being deleted from the population because the individuals own the avr allele mostly in the form of heterozygote at the initial phase after the mutation. Since only few among many models of plant resistance deployment study the case of diploid pathogen and the contribution of the pathogen life cycle, this work will add an important intellect to the literature (Rimbaud et al. 2021).
From the study of host-parasite interaction in flax rust Melampsora lini, Flor (1942, 1955) showed that the host resistance is formed by the interaction of a host resistance gene and a corresponding pathogen gene. This gene-for-gene hypothesis has been supported by experimental evidence and has served as a basis of the methods of molecular breeding targeting the dominant R genes. However, modern agriculture provides the pathogen populations with the homogeneous environments and laid strong selection pressure on them. As a result, the newly developed resistant plants face the risk of immediate resistance breakdown (Möller and Stukenbrock 2017).
Currently, quantitative resistance is getting attention as characters as a potential target for long-life (mild) resistant breeds (Lannou, 2012). They are polygenic and controlled partly by the same genes that mediate qualitative resistance but mostly by the genes that encode defense-related outputs such as strengthening of the cell wall or defense compound biosynthesis (Corwin and Kliebenstein, 2017). Progress of molecular genetics may overcome the technical difficulty (Bakkeren and Szabo, 2020). Saubin et al. (2021) notes that the pattern of genetic inheritance of the pathogen counterparts that respond to the host traits is crucial regarding with the durability of the resistant hosts. The resistance traits for which avr alleles are predicted to be recessive may be the targets of breeding.
References
Bakkeren, G., and Szabo, L. J. (2020) Progress on molecular genetics and manipulation of rust fungi. Phytopathology, 110, 532-543. https://doi.org/10.1094/PHYTO-07-19-0228-IA
Corwin, J. A., and Kliebenstein, D. J. (2017) Quantitative resistance: more than just perception of a pathogen. The Plant Cell, 29, 655-665. https://doi.org/10.1105/tpc.16.00915
Flor, H. H. (1942) Inheritance of pathogenicity in a cross between physiological races 22 and 24 of Melampsova lini. Phytopathology, 35. Abstract.
Flor, H. H. (1955) Host-parasite interactions in flax rust-its genetics and other implications. Phytopathology, 45, 680-685.
Lannou, C. (2012) Variation and selection of quantitative traits in plant pathogens. Annual review of phytopathology, 50, 319-338. https://doi.org/10.1146/annurev-phyto-081211-173031
Möller, M. and Stukenbrock, E. H. (2017) Evolution and genome architecture in fungal plant pathogens. Nature Reviews Microbiology. 15, 756–771. https://doi.org/10.1038/nrmicro.2017.76
Rimbaud, L., Fabre, F., Papaïx, J., Moury, B., Lannou, C., Barrett, L. G., and Thrall, P. H. (2021) Models of Plant Resistance Deployment. Annual Review of Phytopathology, 59. https://doi.org/10.1146/annurev-phyto-020620-122134
Saubin, M., De Mita, S., Zhu, X., Sudret, B. and Halkett, F. (2021) Impact of ploidy and pathogen life cycle on resistance durability. bioRxiv, 2021.05.28.446112, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.05.28.446112
Genome plasticity in Papillomaviruses and de novo emergence of E5 oncogenes
E5, the third oncogene of Papillomavirus
Recommended by Hirohisa Kishino based on reviews by Leonardo de Oliveira Martins and 1 anonymous reviewerPapillomaviruses (PVs) infect almost all mammals and possibly amniotes and bony fishes. While most of them have no significant effects on the hosts, some induce physical lesions. Phylogeny of PVs consists of a few crown groups [1], among which AlphaPVs that infect primates including human have been well studied. They are associated to largely different clinical manifestations: non-oncogenic PVs causing anogenital warts, oncogenic and non-oncogenic PVs causing mucosal lesions, and non-oncogenic PVs causing cutaneous warts.
The PV genome consists of a double stranded circular DNA genome, roughly organized into three parts: an early region coding for six open reading frames (ORFs: E1, E2, E4, E5, E6 and E7) involved in multiple functions including viral replication and cell transformation; a late region coding for structural proteins (L1 and L2); and a non-coding regulatory region (URR) that contains the cis-elements necessary for replication and transcription of the viral genome.
The E5, E6, and E7 are known to act as oncogenes. The E6 protein binds to the cellular p53 protein [2]. The E7 protein binds to the retinoblastoma tumor suppressor gene product, pRB [3]. However, the E5 has been poorly studied, even though a high correlation between the type of E5 protein and the infection phenotype is observed. E5s, being present on the E2/L2 intergenic region in the genomes of a few polyphyletic PV lineages, are so diverged and can only be characterized by high hydrophobicity. No similar sequences have been found in the sequence database.
Willemsen et al. [4] provide valuable evidence on the origin and evolutionary history of E5 genes and their genomic environments. First, they tested common ancestry vs independent origins [5]. Because alignment can lead to biased testing toward the hypothesis of common ancestry [6], they took full account of alignment uncertainty [7] and conducted random permutation test [8]. Although the strong chemical similarity hampered decisive conclusion on the test, they could confirm that E5 may do code proteins, and have unique evolutionary history with far different topology from the neighboring genes.
Still, there is mysteries with the origin and evolution of E5 genes. One of the largest interest may be the evolution of hydrophobicity, because it may be the main cause of variable infection phenotype. The inference has some similarity in nature with the inference of evolutionary history of G+C contents in bacterial genomes [9]. The inference may take account of possible opportunity of convergent or parallel evolution by setting an anchor to the topologies of neighboring genes.
References
[1] Bravo, I. G., & Alonso, Á. (2004). Mucosal human papillomaviruses encode four different E5 proteins whose chemistry and phylogeny correlate with malignant or benign growth. Journal of virology, 78, 13613-13626. doi: 10.1128/JVI.78.24.13613-13626.2004
[2] Werness, B. A., Levine, A. J., & Howley, P. M. (1990). Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science, 248, 76-79. doi: 10.1126/science.2157286
[3] Dyson, N., Howley, P. M., Munger, K., & Harlow, E. D. (1989). The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science, 243, 934-937. doi: 10.1126/science.2537532
[4] Willemsen, A., Félez-Sánchez, M., & Bravo, I. G. (2019). Genome plasticity in Papillomaviruses and de novo emergence of E5 oncogenes. bioRxiv, 337477, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/337477
[5] Theobald, D. L. (2010). A formal test of the theory of universal common ancestry. Nature, 465, 219–222. doi: 10.1038/nature09014
[6] Yonezawa, T., & Hasegawa, M. (2010). Was the universal common ancestry proved?. Nature, 468, E9. doi: 10.1038/nature09482
[7] Redelings, B. D., & Suchard, M. A. (2005). Joint Bayesian estimation of alignment and phylogeny. Systematic biology, 54(3), 401-418. doi: 10.1080/10635150590947041
[8] de Oliveira Martins, L., & Posada, D. (2014). Testing for universal common ancestry. Systematic biology, 63(5), 838-842. doi: 10.1093/sysbio/syu041
[9] Galtier, N., & Gouy, M. (1998). Inferring pattern and process: maximum-likelihood implementation of a nonhomogeneous model of DNA sequence evolution for phylogenetic analysis. Molecular biology and evolution, 15(7), 871-879. doi: 10.1093/oxfordjournals.molbev.a025991