
Latest recommendations

| Id | Title | Authors | Abstract▲ | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
11 Sep 2017

POSTPRINT
Less effective selection leads to larger genomesColonisation of subterranean ecosystems leads to larger genome in waterlouse (Aselloidea)Recommended by Benoit Nabholz and Jochen B. W. WolfThe total amount of DNA utilized to store hereditary information varies immensely among eukaryotic organisms. Single copy genome sizes – disregarding differences due to ploidy - differ by more than three orders of magnitude ranging from a few million nucleotides (Mb) to hundreds of billions (Gb). With the ever-increasing availability of fully sequenced genomes we now know that most of the difference is due either to whole genome duplication or to variation in the abundance of repetitive elements. Regarding repetitive elements, the evolutionary forces underlying the large variation 'allowing' more or less elements in a genome remain largely elusive. A tentative correlation between an organism's complexity (however this may be adequately measured) and genome size, the so called C-value paradox [1], has long been dismissed. Studies testing for selection on secondary phenotypic effects associated with genome size (cell size, metabolic rates, nutrient availability) have yielded mixed results. Nonadaptive theories capitalizing on a role of deleterious insertion-deletion mutations and genetic drift as the main drivers have likewise received mixed support [2-3]. Overall, most evidence was derived from analyses across broad taxonomical scales [4-6]. Lefébure and colleagues [7] take a different approach. They confine their considerations to a homogeneous, restricted taxonomical group, isopod crustaceans of the superfamily Aselloidea. This taxonomic focus allows the authors to circumvent many of the confounding factors such as phylogenetic inertia, life history divergence and mutation rate variation that tend to trouble analyses across broad taxonomic timescales. Another important feature of the chosen system is the evolutionary independent transition of habitat use that has occurred at least 11 times. One group of species inhabits subterranean ecosystems (groundwater), another group thrives on surface water. Populations of the former live in low-energy habitats and are expected to be outnumbered by their surface dwelling relatives. Interestingly – and a precondition for the study - the groundwater species have significantly larger genomes (up to 137%). With this unique set-up, the authors are able to investigate the link between genome size and evolutionary forces related to a proxy of long-term population size by removing many of the confounding factors a priori. Upfront, we learn that the dN/dS ratio is higher in the groundwater species. This may either suggest prevalent positive selection or lower efficacy of purifying selection (relaxed constraint) in the group of species in which population sizes are expected to be low. Using a series of population genetic analyses the authors provide compelling evidence for the latter. Analyses are carefully conducted and include models for estimating the intensity and frequency of purifying and positive selection, the DoS (direction of selection) and α statistic. Next the authors also exclude the possibility that increased dN/dS of the subterranean groundwater species may be due to nonfunctionalization, which may result from the subterranean lifestyle. Overall, these analyses suggest relaxed constraint in smaller populations as the most plausible alternative to explain increased dN/dS ratios. In addition to the efficacy of selection, the authors estimate the timing of the ecological transition under the rationale that the amount of time a species may have been exposed to the subterranean habitat may reflect long term population sizes. To calibrate the 'colonization clock' they apply a neat trick based on the degree of degeneration of the opsin gene (as vision tends to get lost in these habitats). When finally testing which parameters may explain differences in genome size all factors – ecological status, selection efficiency as measured by dN/dS and colonization time - turned out to be significant predictors. Direct estimates of the short term effective population size Ne from polymorphism data, however, did not correlate with genome size. Ruling out the effect of other co-variates such as body size and growth rate the authors conclude that genome size was overall best predicted by long-term population size change upon habitat shift. In that the authors provide convincing evidence that the increase in genome size is linked to a decrease in long-term reduction of selection efficiency of subterranean species. Assuming a bias for insertion mutations over deletion mutations (which is usually the case in eukaryotes) this result is in agreement with the theory of mutational hazard [4-6]. This theory proposed by Michael Lynch postulates that the accumulation of non-functional DNA has a weak deleterious effect that can only be efficiently opposed by natural selection in species with high Ne. In conclusion, Lefébure and colleagues provide novel and welcome evidence supporting a 'neutralist' hypothesis of genome size evolution without the need to invoke an adaptive component. Methodologically, the study cautions against the common use of polymorphism-based estimates of Ne which are often obfuscated by transitory demographic change. Instead, alternative measures of selection efficacy linked to long-term population size may serve as better predictors of genome size. We hope that this study will stimulate additional work testing the link between Ne and genome size variation in other taxonomical groups [8-9]. Using genome sequences instead of the transcriptome approach applied here may concomitantly further our understanding of the molecular mechanisms underlying genome size change. References [1] Thomas, CA Jr. 1971. The genetic organization of chromosomes. Annual Review of Genetics 5: 237–256. doi: 10.1146/annurev.ge.05.120171.001321 [2] Ågren JA, Greiner S, Johnson MTJ, Wright SI. 2015. No evidence that sex and transposable elements drive genome size variation in evening primroses. Evolution 69: 1053–1062. doi: 10.1111/evo.12627 [3] Bast J, Schaefer I, Schwander T, Maraun M, Scheu S, Kraaijeveld K. 2016. No accumulation of transposable elements in asexual arthropods. Molecular Biology and Evolution 33: 697–706. doi: 10.1093/molbev/msv261 [4] Lynch M. 2007. The Origins of Genome Architecture. Sinauer Associates. [5] Lynch M, Bobay LM, Catania F, Gout JF, Rho M. 2011. The repatterning of eukaryotic genomes by random genetic drift. Annual Review of Genomics and Human Genetics 12: 347–366. doi: 10.1146/annurev-genom-082410-101412 [6] Lynch M, Conery JS. 2003. The origins of genome complexity. Science 302: 1401–1404. doi: 10.1126/science.1089370 [7] Lefébure T, Morvan C, Malard F, François C, Konecny-Dupré L, Guéguen L, Weiss-Gayet M, Seguin-Orlando A, Ermini L, Der Sarkissian C, Charrier NP, Eme D, Mermillod-Blondin F, Duret L, Vieira C, Orlando L, and Douady CJ. 2017. Less effective selection leads to larger genomes. Genome Research 27: 1016-1028. doi: 10.1101/gr.212589.116 [8] Lower SS, Johnston JS, Stanger-Hall KF, Hjelmen CE, Hanrahan SJ, Korunes K, Hall D. 2017. Genome size in North American fireflies: Substantial variation likely driven by neutral processes. Genome Biolology and Evolution 9: 1499–1512. doi: 10.1093/gbe/evx097 [9] Sessegolo C, Burlet N, Haudry A. 2016. Strong phylogenetic inertia on genome size and transposable element content among 26 species of flies. Biology Letters 12: 20160407. doi: 10.1098/rsbl.2016.0407 | Less effective selection leads to larger genomes | Tristan Lefébure, Claire Morvan, Florian Malard, Clémentine François, Lara Konecny-Dupré, Laurent Guéguen, Michèle Weiss-Gayet, Andaine Seguin-Orlando, Luca Ermini, Clio Der Sarkissian, N. Pierre Charrier, David Eme, Florian Mermillod-Blondin, Lau... | The evolutionary origin of the striking genome size variations found in eukaryotes remains enigmatic. The effective size of populations, by controlling selection efficacy, is expected to be a key parameter underlying genome size evolution. However... | | Evolutionary Theory, Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Benoit Nabholz | 2017-09-08 09:39:23 | ||
07 Jul 2017

Negative frequency-dependent selection is frequently confoundingUnmasking the delusive appearance of negative frequency-dependent selectionRecommended by Ignacio Bravo based on reviews by David Baltrus and 2 anonymous reviewersExplaining the processes that maintain polymorphisms in a population has been a fundamental line of research in evolutionary biology. One of the main mechanisms identified that preserves genetic diversity is negative frequency-dependent selection (NFDS), which constitutes a powerful framework for interpreting the presence of persistent polymorphisms. Nevertheless, a number of patterns that are often explained by invoking NFDS may also be compatible with, and possibly more easily explained by, different processes. References [1] Brisson D. 2017. Negative frequency-dependent selection is frequently confounding. bioRxiv 113324, ver. 3 of 20th June 2017. doi: 10.1101/113324 [2] Heino M, Metz JAJ and Kaitala V. 1998. The enigma of frequency-dependent selection. Trends in Ecology & Evolution 13: 367-370. doi: 1016/S0169-5347(98)01380-9 | Negative frequency-dependent selection is frequently confounding | Dustin Brisson | The existence of persistent genetic variation within natural populations presents an evolutionary problem as natural selection and genetic drift tend to erode genetic diversity. Models of balancing selection were developed to account for the high ... | | Evolutionary Applications, Evolutionary Theory, Population Genetics / Genomics | Ignacio Bravo | 2017-03-03 18:46:42 | ||
20 Nov 2017
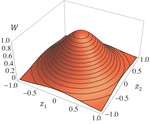
Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selectionUnderstanding genetic variance, load, and inbreeding depression with selfingRecommended by Aneil F. Agrawal based on reviews by Frédéric Guillaume and 1 anonymous reviewerA classic problem in evolutionary biology is to understand the genetic variance in fitness. The simplest hypothesis is that variation exists, even in well-adapted populations, as a result of the balance between mutational input and selective elimination. This variation causes a reduction in mean fitness, known as the mutation load. Though mutation load is difficult to quantify empirically, indirect evidence of segregating genetic variation in fitness is often readily obtained by comparing the fitness of inbred and outbred offspring, i.e., by measuring inbreeding depression. Mutation-selection balance models have been studied as a means of understanding the genetic variance in fitness, mutation load, and inbreeding depression. Since their inception, such models have increased in sophistication, allowing us to ask these questions under more realistic and varied scenarios. The new theoretical work by Abu Awad and Roze [1] is a substantial step forward in understanding how arbitrary levels of self-fertilization affect variation, load and inbreeding depression under mutation-selection balance. References [1] Abu Awad D and Roze D. 2017. Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selection. bioRxiv, 180000, ver. 4 of 17th November 2017. doi: 10.1101/180000 [2] Lande R. 1977. The influence of the mating system on the maintenance of genetic variability in polygenic characters. Genetics 86: 485–498. [3] Charlesworth D and Charlesworth B. 1987. Inbreeding depression and its evolutionary consequences. Annual Review of Ecology and Systematics. 18: 237–268. doi: 10.1111/10.1146/annurev.es.18.110187.001321 [4] Lande R and Porcher E. 2015. Maintenance of quantitative genetic variance under partial self-fertilization, with implications for the evolution of selfing. Genetics 200: 891–906. doi: 10.1534/genetics.115.176693 [5] Roze D. 2015. Effects of interference between selected loci on the mutation load, inbreeding depression, and heterosis. Genetics 201: 745–757. doi: 10.1534/genetics.115.178533 [6] Martin G and Lenormand T. 2006. A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species. Evolution 60: 893–907. doi: 10.1111/j.0014-3820.2006.tb01169.x [7] Martin G, Elena SF and Lenormand T. 2007. Distributions of epistasis in microbes fit predictions from a fitness landscape model. Nature Genetics 39: 555–560. doi: 10.1038/ng1998 | Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selection | Diala Abu Awad and Denis Roze | The mating system of a species is expected to have important effects on its genetic diversity. In this paper, we explore the effects of partial selfing on the equilibrium genetic variance Vg, mutation load L and inbreeding depression δ under stabi... | | Evolutionary Theory, Population Genetics / Genomics, Quantitative Genetics, Reproduction and Sex | Aneil F. Agrawal | 2017-08-26 09:29:20 | ||
20 Dec 2017

Renewed diversification following Miocene landscape turnover in a Neotropical butterfly radiationThe influence of environmental change over geological time on the tempo and mode of biological diversification, revealed by Neotropical butterfliesRecommended by Richard H Ree based on reviews by Delano Lewis and 1 anonymous reviewerThe influence of environmental change over geological time on the tempo and mode of biological diversification is a hot topic in biogeography. Of central interest are questions about where, when, and how fast lineages proliferated, suffered extinction, and migrated in response to tectonic events, the waxing and waning of dominant biomes, etc. In this context, the dynamic conditions of the Miocene have received much attention, from studies of many clades and biogeographic regions. Here, Chazot et al. [1] present an exemplary analysis of butterflies (tribe Ithomiini) in the Neotropics, examining their diversification across the Andes and Amazon. They infer sharp contrasts between these regions in the late Miocene: accelerated diversification during orogeny of the Andes, and greater extinction in the Amazon associated during the Pebas system, with interchange and local diversification increasing following the Pebas during the Pliocene. References [1] Chazot N, Willmott KR, Lamas G, Freitas AVL, Piron-Prunier F, Arias CF, Mallet J, De-Silva DL and Elias M. 2017. Renewed diversification following Miocene landscape turnover in a Neotropical butterfly radiation. BioRxiv 148189, ver 4 of 19th December 2017. doi: 10.1101/148189 [2] Xing Y, and Ree RH. 2017. Uplift-driven diversification in the Hengduan Mountains, a temperate biodiversity hotspot. Proceedings of the National Academy of Sciences of the United States of America, 114: E3444-E3451. doi: 10.1073/pnas.1616063114 | Renewed diversification following Miocene landscape turnover in a Neotropical butterfly radiation | Nicolas Chazot, Keith R. Willmott, Gerardo Lamas, André V.L. Freitas, Florence Piron-Prunier, Carlos F. Arias, James Mallet, Donna Lisa De-Silva, Marianne Elias | The Neotropical region has experienced a dynamic landscape evolution throughout the Miocene, with the large wetland Pebas occupying western Amazonia until 11-8 my ago and continuous uplift of the Andes mountains along the western edge of South Ame... | | Macroevolution, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Richard H Ree | 2017-06-12 11:55:14 | ||
17 Dec 2016
POSTPRINT
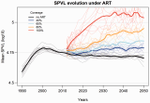
Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling studyPredicting HIV virulence evolution in response to widespread treatmentRecommended by Samuel Alizon and Roger Kouyos and Roger Kouyos
It is a classical result in the virulence evolution literature that treatments decreasing parasite replication within the host should select for higher replication rates, thus driving increased levels of virulence if the two are correlated. There is some evidence for this in vitro but very little in the field. HIV infections in humans offer a unique opportunity to go beyond the simple predictions that treatments should favour more virulent strains because many details of this host-parasite system are known, especially the link between set-point virus load, transmission rate and virulence. To tackle this question, Herbeck et al. [1] used a detailed individual-based model. This is original because it allows them to integrate existing knowledge from the epidemiology and evolution of HIV (e.g. recent estimates of the ‘heritability’ of set-point virus load from one infection to the next). This detailed model allows them to formulate predictions regarding the effect of different treatment policies; especially regarding the current policy switch away from treatment initiation based on CD4 counts towards universal treatment. The results show that, perhaps as expected from the theory, treatments based on the level of remaining host target cells (CD4 T cells) do not affect virulence evolution because they do not strongly affect the virulence level that maximizes HIV’s transmission potential. However, early treatments can lead to moderate increase in virulence within several years if coverage is high enough. These results seem quite robust to variation of all the parameters in realistic ranges. The great step forward in this model is the ability to obtain quantitative prediction regarding how a virus may evolve in response to public health policies. Here the main conclusion is that given our current knowledge in HIV biology, the risk of virulence evolution is perhaps more limited than expected from a direct application of virulence evolution model. Interestingly, the authors also conclude that recently observed increased in HIV virulence [2-3] cannot be explained by the impact of antiretroviral therapy alone; which raises the question about the main mechanism behind this increase. Finally, the authors make the interesting suggestion that “changing virulence is amenable to being monitored alongside transmitted drug resistance in sentinel surveillance”. References [1] Herbeck JT, Mittler JE, Gottlieb GS, Goodreau SM, Murphy JT, Cori A, Pickles M, Fraser C. 2016. Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling study. Virus Evolution 2:vew028. doi: 10.1093/ve/vew028 [2] Herbeck JT, Müller V, Maust BS, Ledergerber B, Torti C, et al. 2012. Is the virulence of HIV changing? A meta-analysis of trends in prognostic markers of HIV disease progression and transmission. AIDS 26:193-205. doi: 10.1097/QAD.0b013e32834db418 [3] Pantazis N, Porter K, Costagliola D, De Luca A, Ghosn J, et al. 2014. Temporal trends in prognostic markers of HIV-1 virulence and transmissibility: an observational cohort study. Lancet HIV 1:e119-26. doi: 10.1016/s2352-3018(14)00002-2 | Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling study | Herbeck JT, Mittler JE, Gottlieb GS, Goodreau SM, Murphy JT, Cori A, Pickles M, Fraser C | There are global increases in the use of HIV antiretroviral therapy (ART), guided by clinical benefits of early ART initiation and the efficacy of treatment as prevention of transmission. Separately, it has been shown theoretically and empirically... | | Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Epidemiology | Samuel Alizon | 2016-12-16 20:54:08 | ||
16 Dec 2016

POSTPRINT
Evolutionary robotics simulations help explain why reciprocity is rare in nature.Simulated robots and the evolution of reciprocityRecommended by Michael D Greenfield and Joël Meunier
Of the various forms of cooperative and altruistic behavior, reciprocity remains the most contentious. Humans certainly exhibit reciprocity – under certain circumstances – and various non-human animals behave in ways suggesting that they do as well. Thus, evolutionary biologists have sought to explain why non-relatives might engage in altruistic transactions when a substantial delay occurs between helping and compensation; i.e. an individual may be a donor today and a beneficiary tomorrow, or vice-versa. This quest, aided by game theory and computer modeling late in the past century, identified some strategies for reciprocal behavior that could work – in theory. But when biologists looked for confirmation of these strategies in animals they found little evidence that stood up to rigorous testing. In a recent paper André and Nolfi [1] offer a compelling reason for this observed rarity of reciprocity: Reciprocal behavior that animals might exhibit is a bit more complex than any of the game theoretic strategies, and even the simplest forms of realistic behavior would entail several nearly simultaneous mutations, an unlikely occurrence. André and Nolfi [1] relied on neural networks to test actors, robots that could evolve helping and reciprocal behavior from a basal level of selfishness. In an extensive series of simulations, they found that reciprocal behavior did not take hold in a population, largely because the various intermediates to full reciprocity were eliminated before the subsequent mutations occurred. The findings are satisfying given our current knowledge of animal behavior, but questions remain. Notably, how does one account for those rare cases in which reciprocity does meet all the criteria? The authors suggest some possibilities, but an analysis will await their next study. Reference [1] André J-B, Nolfi S. 2016. Evolutionary robotics simulations help explain why reciprocity is rare in nature. Scientific Reports 6:32785. doi: 10.1038/srep32785 | Evolutionary robotics simulations help explain why reciprocity is rare in nature. | André J-B, Nolfi S | The relative rarity of reciprocity in nature, contrary to theoretical predictions that it should be widespread, is currently one of the major puzzles in social evolution theory. Here we use evolutionary robotics to solve this puzzle. We show tha... | | Behavior & Social Evolution, Evolutionary Theory | Michael D Greenfield | 2016-12-16 18:08:31 | ||
14 Mar 2017

POSTPRINT
Evolution of multiple sensory systems drives novel egg-laying behavior in the fruit pest Drosophila suzukiiA valuable work lying at the crossroad of neuro-ethology, evolution and ecology in the fruit pest Drosophila suzukiiRecommended by Arnaud Estoup and Ruth Arabelle HufbauerAdaptations to a new ecological niche allow species to access new resources and circumvent competitors and are hence obvious pathways of evolutionary success. The evolution of agricultural pest species represents an important case to study how a species adapts, on various timescales, to a novel ecological niche. Among the numerous insects that are agricultural pests, the ability to lay eggs (or oviposit) in ripe fruit appears to be a recurrent scenario. Fruit flies (family Tephritidae) employ this strategy, and include amongst their members some of the most destructive pests (e.g., the olive fruit fly Bactrocera olea or the medfly Ceratitis capitata). In their ms, Karageorgi et al. [1] studied how Drosophila suzukii, a new major agricultural pest species that recently invaded Europe and North America, evolved the novel behavior of laying eggs into undamaged fresh fruit. The close relatives of D. suzukii lay their eggs on decaying plant substrates, and thus this represents a marked change in host use that links to substantial economic losses to the fruit industry. Although a handful of studies have identified genetic changes causing new behaviors in various species, the question of the evolution of behavior remains a largely uncharted territory. The study by Karageorgi et al. [1] represents an original and most welcome contribution in this domain for a non-model species. Using clever behavioral experiments to compare D. suzukii to several related Drosophila species, and complementing those results with neurogenetics and mutant analyses using D. suzukii, the authors nicely dissect the sensory changes at the origin of the new egg-laying behavior. The experiments they describe are easy to follow, richly illustrate through figures and images, and particularly well designed to progressively decipher the sensory bases driving oviposition of D. suzukii on ripe fruit. Altogether, Karageorgi et al.’s [1] results show that the egg-laying substrate preference of D. suzukii has considerably evolved in concert with its morphology (especially its enlarged, serrated ovipositor that enables females to pierce the skin of many ripe fruits). Their observations clearly support the view that the evolution of traits that make D. suzukii an agricultural pest included the modification of several sensory systems (i.e. mechanosensation, gustation and olfaction). These differences between D. suzukii and its close relatives collectively underlie a radical change in oviposition behavior, and were presumably instrumental in the expansion of the ecological niche of the species. The authors tentatively propose a multi-step evolutionary scenario from their results with the emergence of D. suzukii as a pest species as final outcome. Such formalization represents an interesting evolutionary model-framework that obviously would rely upon further data and experiments to confirm and refine some of the evolutionary steps proposed, especially the final and recent transition of D. suzukii from non-invasive to invasive species. References [1] Karageorgi M, Bräcker LB, Lebreton S, Minervino C, Cavey M, Siju KP, Grunwald Kadow IC, Gompel N, Prud’homme B. 2017. Evolution of multiple sensory systems drives novel egg-laying behavior in the fruit pest Drosophila suzukii. Current Biology, 27: 1-7. doi: 10.1016/j.cub.2017.01.055 | Evolution of multiple sensory systems drives novel egg-laying behavior in the fruit pest Drosophila suzukii | Marianthi Karageorgi, Lasse B. Bräcker, Sébastien Lebreton, Caroline Minervino, Matthieu Cavey, K.P. Siju, Ilona C. Grunwald Kadow, Nicolas Gompel, Benjamin Prud’homme | The rise of a pest species represents a unique opportunity to address how species evolve new behaviors and adapt to novel ecological niches. We address this question by studying the egg-laying behavior of Drosophila suzukii, an invasive agricultur... | | Adaptation, Behavior & Social Evolution, Evo-Devo, Evolutionary Applications, Evolutionary Ecology, Expression Studies, Genotype-Phenotype, Macroevolution, Molecular Evolution | Arnaud Estoup | 2017-03-13 17:42:00 | ||
13 Dec 2016

POSTPRINT
Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax)Supergene Control of a Reproductive PolymorphismRecommended by Thomas Flatt and Laurent KellerTwo back-to-back papers published earlier this year in Nature Genetics provide compelling evidence for the control of a male reproductive polymorphism in a wading bird by a "supergene", a cluster of tightly linked genes [1-2]. The bird in question, the ruff (Philomachus pugnax), has a rather unusual reproductive system that consists of three distinct types of males ("reproductive morphs"): aggressive "independents" who represent the majority of males; a smaller fraction of non-territorial "satellites" who are submissive towards "independents"; and "faeders" who mimic females and are rare. Previous work has shown that the male morphs differ in major aspects of mating and aggression behavior, plumage coloration and body size, and that – intriguingly – this complex multi-trait polymorphism is apparently controlled by a single autosomal Mendelian locus with three alleles [3]. To uncover the genetic control of this polymorphism two independent teams, led by Terry Burke [1] and Leif Andersson [2], have set out to analyze the genomes of male ruffs. Using a combination of genomics and genetics, both groups managed to pin down the supergene locus and map it to a non-recombining, 4.5 Mb large inversion which arose 3.8 million years ago. While "independents" are homozygous for the ancestral uninverted sequence, "satellites" and "faeders" carry evolutionarily divergent, dominant alternative haplotypes of the inversion. Thus, as in several other notable cases, for example the supergene control of disassortative mating, aggressiveness and plumage color in white-throated sparrows [4], of mimicry in Heliconius and Papilio butterflies [5-6], or of social structure in ants [7], an inversion – behaving as a single "locus" – underpins the mechanistic basis of the supergene. More generally, and beyond inversions, a growing number of studies now shows that selection can favor the evolution of suppressed recombination, thereby leading to the emergence of clusters of tightly linked loci which can then control – presumably due to polygenic gene action – a suite of complex phenotypes [8-10]. A largely unresolved question in this field concerns the identity of the causative alleles and loci within a given supergene. Recent progress on this question has been made for example in Papilio polytes butterflies where a mimicry supergene has been found to involve – surprisingly – only a single but large gene: multiple mimicry alleles in the doublesex gene are maintained in strong linkage disequilibrium via an inversion. It will clearly be of great interest to see future examples of such a fine-scale genetic dissection of supergenes. In conclusion, we were impressed by the data and analyses of Küpper et al. [1] and Lamichhaney et al. [2]: both papers beautifully illustrate how genomics and evolutionary ecology can be combined to make new, exciting discoveries. Both papers will appeal to readers with an interest in supergenes, inversions, the interplay of selection and recombination, or the genetic control of complex phenotypes. References [1] Küpper C, Stocks M, Risse JE, dos Remedios N, Farrell LL, McRae SB, Morgan TC, Karlionova N, Pinchuk P, Verkuil YI, et al. 2016. A supergene determines highly divergent male reproductive morphs in the ruff. Nature Genetics 48:79-83. doi: 10.1038/ng.3443 [2] Lamichhaney S, Fan G, Widemo F, Gunnarsson U, Thalmann DS, Hoeppner MP, Kerje S, Gustafson U, Shi C, Zhang H, et al. 2016. Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax). Nature Genetics 48:84-88. doi: 10.1038/ng.3430 [3] Lank DB, Smith CM, Hanotte O, Burke T, Cooke F. 1995. Genetic polymorphism for alternative mating behaviour in lekking male ruff Philomachus pugnax. Nature 378:59-62. doi: 10.1038/378059a0 [4] Tuttle Elaina M, Bergland Alan O, Korody Marisa L, Brewer Michael S, Newhouse Daniel J, Minx P, Stager M, Betuel A, Cheviron Zachary A, Warren Wesley C, et al. 2016. Divergence and Functional Degradation of a Sex Chromosome-like Supergene. Current Biology 26:344-350. doi: 10.1016/j.cub.2015.11.069 [5] Joron M, Frezal L, Jones RT, Chamberlain NL, Lee SF, Haag CR, Whibley A, Becuwe M, Baxter SW, Ferguson L, et al. 2011. Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477:203-206. doi: 10.1038/nature10341 [6] Kunte K, Zhang W, Tenger-Trolander A, Palmer DH, Martin A, Reed RD, Mullen SP, Kronforst MR. 2014. doublesex is a mimicry supergene. Nature 507:229-232. doi: 10.1038/nature13112 [7] Wang J, Wurm Y, Nipitwattanaphon M, Riba-Grognuz O, Huang Y-C, Shoemaker D, Keller L. 2013. A Y-like social chromosome causes alternative colony organization in fire ants. Nature 493:664-668. doi: 10.1038/nature11832 [8] Thompson MJ, Jiggins CD. 2014. Supergenes and their role in evolution. Heredity 113:1-8. doi: 10.1038/hdy.2014.20 [9] Schwander T, Libbrecht R, Keller L. 2014. Supergenes and Complex Phenotypes. Current Biology 24:R288-R294. doi: 10.1016/j.cub.2014.01.056 [10] Charlesworth D. 2015. The status of supergenes in the 21st century: recombination suppression in Batesian mimicry and sex chromosomes and other complex adaptations. Evolutionary Applications 9:74-90. doi: 10.1111/eva.12291 | Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax) | Lamichhaney S, Fan G, Widemo F, Gunnarsson U, Thalmann DS, Hoeppner MP, Kerje S, Gustafson U, Shi C, Zhang H, et al. | The ruff is a Palearctic wader with a spectacular lekking behavior where highly ornamented males compete for females1, 2, 3, 4. This bird has one of the most remarkable mating systems in the animal kingdom, comprising three different male morphs (... | | Adaptation, Behavior & Social Evolution, Genotype-Phenotype, Life History, Population Genetics / Genomics, Quantitative Genetics, Reproduction and Sex | Thomas Flatt | 2016-12-13 17:46:54 | ||
12 Jul 2017

Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategiesTowards an integrated scenario to understand evolutionary patterns in A. thalianaRecommended by Xavier Picó based on reviews by Rafa Rubio de Casas and Xavier PicóNobody can ignore that a full understanding of evolution requires an integrated approach from both conceptual and methodological viewpoints. Although some life-history traits, e.g. flowering time, have long been receiving more attention than others, in many cases because the former are more workable than the latter, we must acknowledge that our comprehension about how evolution works is strongly biased and limited. In the Arabidopsis community, such an integration is making good progress as an increasing number of research groups worldwide are changing the way in which evolution is put to the test. This manuscript [1] is a good example of that as the authors raise an important issue in evolutionary biology by combining gene expression and flowering time data from different sources. In particular, the authors explore how variation in flowering time, which determines lifespan, and host immunity defenses co-vary, which is interpreted in terms of co-evolution between the two traits. Interestingly, the authors go beyond that pattern by separating lifespan-dependent from lifespan–independent defense genes, and by showing that defense genes with variants known to impact fitness in the field are among the genes whose expression co-varies most strongly with flowering time. Finally, these results are supported by a simple mathematical model indicating that such a relationship can also be expected theoretically. Overall, the readers will find many conceptual and methodological elements of interest in this manuscript. The idea that evolution is better understood under the scope of life history variation is really exciting and challenging, and in my opinion on the right track for disentangling the inherent complexities of evolutionary research. However, only when we face complexity, we also face its costs and burdens. In this particular case, the well-known co-variation between seed dormancy and flowering time is a missing piece, as well as the identification of (variation in) putative selective pressures accounting for the co-evolution between defense mechanisms and life history (seed dormancy vs. flowering time) along environmental gradients. More intellectual, technical and methodological challenges that with no doubt are totally worth it. Reference [1] Glander S, He F, Schmitz G, Witten A, Telschow A, de Meaux J. 2017. Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategies. bioRxiv ver.1 of June 19, 2017. doi: 10.1101/131136 | Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategies | Glander S, He F, Schmitz G, Witten A, Telschow A, de Meaux J | The selective impact of pathogen epidemics on host defenses can be strong but remains transient. By contrast, life-history shifts can durably and continuously modify the balance between costs and benefits of immunity, which arbitrates the evolutio... | | Adaptation, Evolutionary Ecology, Expression Studies, Life History, Phenotypic Plasticity, Quantitative Genetics, Species interactions | Xavier Picó | Sophie Karrenberg, Rafa Rubio de Casas, Xavier Picó | 2017-06-21 10:57:14 | |
12 Jun 2017

Evolution and manipulation of vector host choiceModelling the evolution of how vector-borne parasites manipulate the vector's host choiceRecommended by Samuel Alizon based on reviews by Samuel Alizon and Nicole Mideo
Many parasites can manipulate their hosts, thus increasing their transmission to new hosts [1]. This is particularly the case for vector-borne parasites, which can alter the feeding behaviour of their hosts. However, predicting the optimal strategy is not straightforward because three actors are involved and the interests of the parasite may conflict with that of the vector. There are few models that consider the evolution of host manipulation by parasites [but see 2-4], but there are virtually none that investigated how parasites can manipulate the host choice of vectors. Even on the empirical side, many aspects of this choice remain unknown. Gandon [5] develops a simple evolutionary epidemiology model that allows him to formulate clear and testable predictions. These depend on which actor controls the trait (the vector or the parasite) and, when there is manipulation, whether it is realised via infected hosts (to attract vectors) or infected vectors (to change host choice). In addition to clarifying the big picture, Gandon [5] identifies some nice properties of the model, for instance an independence of the density/frequency-dependent transmission assumption or a backward bifurcation at R0=1, which suggests that parasites could persist even if their R0 is driven below unity. Overall, this study calls for further investigation of the different scenarios with more detailed models and experimental validation of general predictions. References [1] Hughes D, Brodeur J, Thomas F. 2012. Host manipulation by parasites. Oxford University Press. [2] Brown SP. 1999. Cooperation and conflict in host-manipulating parasites. Proceedings of the Royal Society of London B: Biological Sciences 266: 1899–1904. doi: 10.1098/rspb.1999.0864 [3] Lion S, van Baalen M, Wilson WG. 2006. The evolution of parasite manipulation of host dispersal. Proceedings of the Royal Society of London B: Biological Sciences. 273: 1063–1071. doi: 10.1098/rspb.2005.3412 [4] Vickery WL, Poulin R. 2010. The evolution of host manipulation by parasites: a game theory analysis. Evolutionary Ecology 24: 773–788. doi: 10.1007/s10682-009-9334-0 [5] Gandon S. 2017. Evolution and manipulation of vector host choice. bioRxiv 110577, ver. 3 of 7th June 2017. doi: 10.1101/110577 | Evolution and manipulation of vector host choice | Sylvain Gandon | The transmission of many animal and plant diseases relies on the behavior of arthropod vectors. In particular, the choice to feed on either infected or uninfected hosts can dramatically affect the epidemiology of vector-borne diseases. I develop a... | | Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory | Samuel Alizon | 2017-03-03 19:18:54 |




