
KEMPPAINEN Petri
- Organismal and Evolutionary Biology Research Programme, Eco-Evolutionary Dynamics group, Helsinki, Finland
- Adaptation, Bioinformatics & Computational Biology, Evolutionary Ecology, Hybridization / Introgression, Population Genetics / Genomics, Quantitative Genetics, Speciation
Recommendations: 0
Review: 1
Review: 1
On the potential for GWAS with phenotypic population means and allele-frequency data (popGWAS)
popGWAS: Data-efficient trait mapping in natural populations for biodiversity research
Recommended by Frédéric Guillaume based on reviews by Petri Kemppainen and 1 anonymous reviewerThe study by Pfenninger (2025) addresses the critical need to understand the genomic basis of ecologically important traits to better predict and respond to the impacts of global change on biodiversity (Gienapp et al. 2017). It introduces the popGWAS, a novel GWAS approach, which utilizes phenotypic population means and genome-wide allele frequency data, obtainable through methods like Pool-sequencing (Pool-Seq), to identify the genetic loci underlying quantitative polygenic traits in natural populations and predict their mean. The core idea is that trait-increasing alleles should exhibit higher frequencies in populations with higher mean trait values. popGWAS then maps mean allele frequencies across populations to their trait means. Working with as many allele frequency values as populations sampled, popGWAS potentially has more power to find significant associations at genomic loci than individual-based GWAS working with three genotypes at a locus. This new method addresses some of the problems faced by traditional genome-wide association studies (GWAS), which require extensive resources and large sample sizes, posing challenges for biodiversity research on non-model species in natural populations.
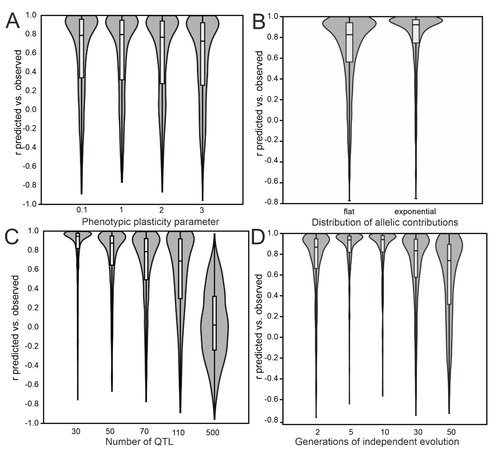
To evaluate the effectiveness of popGWAS, Pfenninger (2025) conducted extensive population genetic forward simulations, examining scenarios with varying numbers of populations, ranging from 12 to 60. The results indicated that popGWAS performance improved with increasing sample size, showing a diminishing return above 36 populations. In a direct comparison across all simulation scenarios, popGWAS consistently outperformed individual-based GWAS (iGWAS). On average, popGWAS identified more true positive loci than iGWAS. In addition, when combined with minimum entropy feature selection (MEFS), popGWAS achieved large predictive accuracy of population means of 0.8 or better in over 97% of simulations with 36 or more populations, regardless of other parameters. In contrast, iGWAS failed to generate valid phenotypic predictions in over 70% of the simulations. Also, unlike iGWAS, popGWAS did not suffer from p-value inflation. Yet, population structure or varying levels of relatedness among individuals were not fully accounted for in the simulations. The extent to which popGWAS would be sensitive to such individual covariates remains to be shown. Finally, popGWAS was relatively insensitive to low trait heritability because random individual variation gets averaged out when calculating the population mean trait value.
The study demonstrates that popGWAS is a promising approach, particularly for oligogenic and moderately polygenic traits. The method performs more poorly for polygenic traits with large genetic redundancy, where different alleles contribute to the same trait mean in different populations. The method thus performs better when large-effect loci contribute to genetic differentiation in parallel across populations, as expected when gene flow is moderate to high (Yeaman & Whitlock 2011). Low genomic predictability is reached when drift dominates or when genetic architectures are highly polygenic.
The popGWAS method proved effective with a moderate number of sampled populations and, when combined with machine learning for genomic prediction, exhibited strong performance in predicting population means, even for low-heritability traits. Notably, popGWAS consistently outperformed iGWAS in terms of identifying true positive loci and prediction accuracy. This suggests that popGWAS can make GWAS studies more accessible for biodiversity genomics research, providing a valuable tool for dissecting the genetic basis of complex traits in natural populations. A key aspect contributing to the efficiency of popGWAS is its compatibility with pooled sequencing (Pool-Seq). Pool-Seq provides estimates of allele frequencies within a population by sequencing a mixed DNA sample representing multiple individuals from that population (Futschik & Schlötterer 2010). This approach is significantly more cost-effective than sequencing each individual separately, allowing researchers to obtain genome-wide allele frequency data across multiple populations with a substantially reduced budget. This data efficiency makes GWAS more accessible to a wider range of researchers, particularly those working in biodiversity genomics where financial resources may be limited. Furthermore, popGWAS can be coupled with bulk phenotyping methods, such as automatic video recording, remote sensing, metabolomics/transcriptomics, etc., to efficiently obtain population-level phenotypic data, further streamlining the research process. Ultimately, popGWAS represents a valuable addition to the geneticist's toolkit, offering a complementary approach to iGWAS that can be particularly advantageous in specific research contexts where predicting trait mean is more important than resolving the precise genetic basis of a trait.
References
Futschik, A. and Schlötterer, C. 2010. The Next Generation of Molecular Markers From Massively Parallel Sequencing of Pooled DNA Samples. Genetics 186(1): 207-218. https://doi.org/10.1534/genetics.110.114397
Gienapp, P., Fior, S., Guillaume, F., Lasky, J. R., Sork, V. L. and Csilléry, K. 2017. Genomic Quantitative Genetics to Study Evolution in the Wild. Trends Ecol. Evol. 32(12): 897-908. https://doi.org/10.1016/j.tree.2017.09.004
Markus Pfenninger (2025) On the potential for GWAS with phenotypic population means and allele-frequency data (popGWAS). bioRxiv, ver.3 peer-reviewed and recommended by PCI Evol Biol https://doi.org/10.1101/2024.06.12.598621
Yeaman, S. and Whitlock, M. C. 2011. The genetic architecture of adaptation under migration-selection balance. Evolution 65(7): 1897-1911. https://doi.org/10.1111/j.1558-5646.2011.01269.x