
Latest recommendations

| Id | Title | Authors▲ | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
25 Jan 2024

Sperm production and allocation in response to risk of sperm competition in the black soldier fly Hermetia illucensElevated sperm production and faster transfer: plastic responses to the risk of sperm competition in males of the black sodier fly Hermetia illuceRecommended by Trine Bilde based on reviews by Rebecca Boulton, Isabel Smallegange and 1 anonymous reviewer based on reviews by Rebecca Boulton, Isabel Smallegange and 1 anonymous reviewer
In this paper (Manas et al., 2023), the authors investigate male responses to risk of sperm competition in the black soldier fly Hermetia illuce, a widespread insect that has gained recent attention for its potential to be farmed for sustainable food production (Tomberlin & van Huis, 2020). Using an experimental approach that simulated low-risk (males were kept individually) and high-risk (males were kept in groups of 10) of sperm competition, they found that males reared in groups showed a significant increase in sperm production compared with males reared individually. This shows a response to the rearing environment in sperm production that is consistent with an increase in the perceived risk of sperm competition. These males were then used in mating experiments to determine whether sperm allocation to females during mating was influenced by the perceived risk of sperm competition. Mating experiments were initiated in groups, since mating only occurs when more than one male and one female are present, indicating strong sexual selection in the wild. Once a copulation began, the pair was moved to a new environment with no competition, with male competitors, or with other females, to test how social environment and potentially the sex of surrounding individuals influenced sperm allocation during mating. Copulation duration and the number of sperm transferred were subsequently counted. In these mating experiments, the number of sperm stored in the female spermathecae increased under immediate risk of sperm competition. Interestingly, this was not because males copulated for longer depending on the risk of sperm competition, indicating that males respond plastically to the risk of competition by elevating their investment in sperm production and speed of sperm transfer. There was no difference between competitive environments consisting of males or females respectively, suggesting that it is the presence of other flies per se that influence sperm allocation. The study provides an interesting new example of how males alter reproductive investment in response to social context and sexual competition in their environment. In addition, it provides new insights into the reproductive biology of the black soldier fly Hermetia illucens, which may be relevant for optimizing farming conditions. References Manas F, Labrousse C, Bressac C (2023) Sperm production and allocation in response to risks of sperm competition in the black soldier fly Hermetia illucens. bioRxiv, 2023.06.20.544772, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.06.20.544772 Tomberlin JK, Van Huis A (2020) Black soldier fly from pest to ‘crown jewel’ of the insects as feed industry: an historical perspective. Journal of Insects as Food and Feed, 6, 1–4. https://doi.org/10.3920/JIFF2020.0003 | Sperm production and allocation in response to risk of sperm competition in the black soldier fly Hermetia illucens | Frédéric Manas, Carole Labrousse, Christophe Bressac | <p style="text-align: justify;">In polyandrous species, competition between males for offspring paternity goes on after copulation through the competition of their ejaculates for the fertilisation of female's oocytes. Given that males allocating m... | | Reproduction and Sex, Sexual Selection | Trine Bilde | 2023-06-26 09:41:07 | ||
13 Nov 2017

Epidemiological trade-off between intra- and interannual scales in the evolution of aggressiveness in a local plant pathogen populationThe pace of pathogens’ adaptation to their host plantsRecommended by Benoit Moury based on reviews by Benoit Moury and 1 anonymous reviewerBecause of their shorter generation times and larger census population sizes, pathogens are usually ahead in the evolutionary race with their hosts. The risks linked to pathogen adaptation are still exacerbated in agronomy, where plant and animal populations are not freely evolving but depend on breeders and growers, and are usually highly genetically homogeneous. As a consequence, the speed of pathogen adaptation is crucial for agriculture sustainability. Unraveling the time scale required for pathogens’ adaptation to their hosts would notably greatly improve our estimation of the risks of pathogen emergence, the efficiency of disease control strategies and the design of epidemiological surveillance schemes. However, the temporal scale of pathogen evolution has received much less attention than its spatial scale [1]. In their study of a wheat fungal disease, Suffert et al. [2] reached contrasting conclusions about the pathogen adaptation depending on the time scale (intra- or inter-annual) and on the host genotype (sympatric or allopatric) considered, questioning the experimental assessment of this important problem. Suffert et al. [2] sampled two pairs of Zymoseptoria tritici (the causal agent of septoria leaf blotch) sub-populations in a bread wheat field plot, representing (i) isolates collected at the beginning or at the end of an epidemic in a single growing season (2009-2010 intra-annual sampling scale) and (ii) isolates collected from plant debris at the end of growing seasons in 2009 and in 2015 (inter-annual sampling scale). Then, they measured in controlled conditions two aggressiveness traits of the isolates of these four Z. tritici sub-populations, the latent period and the lesion size on leaves, on two wheat cultivars. One of the cultivars was considered as "sympatric" because it was at the source of the studied isolates and was predominant in the growing area before the experiment, whereas the other cultivar was considered as "allopatric" since it replaced the previous one and became predominant in the growing area during the sampling period. On the sympatric host, at the intra-annual scale, they observed a marginally-significant decrease in latent period and a significant decrease of the between-isolate variance for this trait, which are consistent with a selection of pathogen variants with an enhanced aggressiveness. In contrast, at the inter-annual scale, no difference in the mean or variance of aggressiveness trait values was observed on the sympatric host, suggesting a lack of pathogen adaptation. They interpreted the contrast between observations at the two time scales as the consequence of a trade-off for the pathogen between a gain of aggressiveness after several generations of asexual reproduction at the intra-annual scale and a decrease of the probability to reproduce sexually and to be transmitted from one growing season to the next. Indeed, at the end of the growing season, the most aggressive isolates are located on the upper leaves of plants, where the pathogen density and hence probably also the probability to reproduce sexually, is lower. On the allopatric host, the conclusion about the pathogen stability at the inter-annual scale was somewhat different, since a significant increase in the mean lesion size was observed (isolates corresponding to the intra-annual scale were not checked on the allopatric host). This shows the possibility for the pathogen to evolve at the inter-annual scale, for a given aggressiveness trait and on a given host. In conclusion, Suffert et al.’s [2] study emphasizes the importance of the experimental design in terms of sampling time scale and host genotype choice to analyze the pathogen adaptation to its host plants. It provides also an interesting scenario, at the crossroad of the pathogen’s reproduction regime, niche partitioning and epidemiological processes, to interpret these contrasted results. Pathogen adaptation to plant cultivars with major-effect resistance genes is usually fast, including in the wheat-Z. tritici system [3]. Therefore, this study will be of great help for future studies on pathogen adaptation to plant partial resistance genes and on strategies of deployment of such resistance at the landscape scale. References [2] Suffert F, Goyeau H, Sache I, Carpentier F, Gelisse S, Morais D and Delestre G. 2017. Epidemiological trade-off between intra- and interannual scales in the evolution of aggressiveness in a local plant pathogen population. bioRxiv, 151068, ver. 3 of 12th November 2017. doi: 10.1101/151068 [3] Brown JKM, Chartrain L, Lasserre-Zuber P and Saintenac C. 2015. Genetics of resistance to Zymoseptoria tritici and applications to wheat breeding. Fungal Genetics and Biology, 79: 33–41. doi: 10.1016/j.fgb.2015.04.017 | Epidemiological trade-off between intra- and interannual scales in the evolution of aggressiveness in a local plant pathogen population | Frederic Suffert, Henriette Goyeau, Ivan Sache, Florence Carpentier, Sandrine Gelisse, David Morais, Ghislain Delestre | The efficiency of plant resistance to fungal pathogen populations is expected to decrease over time, due to its evolution with an increase in the frequency of virulent or highly aggressive strains. This dynamics may differ depending on the scale i... | | Adaptation, Evolutionary Applications, Evolutionary Epidemiology | Benoit Moury | 2017-06-23 21:04:54 | ||
10 Jul 2019
Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pestThe scandalous pestRecommended by Nicolas Galtier based on reviews by 2 anonymous reviewersKoutsovoulos et al. [1] have generated and analysed the first population genomic dataset in root-knot nematode Meloidogyne incognita. Why is this interesting? For two major reasons. First, M. incognita has been documented to be apomictic, i.e., to lack any form of sex. This is a trait of major evolutionary importance, with implications on species adaptive potential. The study of genome evolution in asexuals is fascinating and has the potential to inform on the forces governing the evolution of sex and recombination. Even small amounts of sex, however, are sufficient to restore most of the population genetic properties of true sexuals [2]. Because rare events of sex can remain undetected in the field, to confirm asexuality in M. incognita using genomic data is an important step. The second reason why M. incognita is of interest is that this nematode is one of the most harmful pests currently living on earth. M. incognita feeds on the roots of many cultivated plants, including tomato, bean, and cotton, and has been of major agricultural importance for decades. A number of races were defined based on host specificity. These have played a key role in attempts to control the dynamic of M. incognita populations via crop rotations. Races and management strategies so far lack any genetic basis, hence the second major interest of this study. References [1] Koutsovoulos, G. D., Marques, E., Arguel, M. J., Duret, L., Machado, A. C. Z., Carneiro, R. M. D. G., Kozlowski, D. K., Bailly-Bechet, M., Castagnone-Sereno, P., Albuquerque, E. V., & Danchin, E. G. J. (2019). Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pest. bioRxiv, 362129, ver. 5, peer-reviewed and recommended by Peer Community in Evolutionary Biology. doi: 10.1101/362129 | Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pest | Georgios D. Koutsovoulos, Eder Marques, Marie-Jeanne Arguel, Laurent Duret, Andressa C.Z. Machado, Regina M.D.G. Carneiro, Djampa K. Kozlowski, Marc Bailly-Bechet, Philippe Castagnone-Sereno, Erika V.S. Albuquerque, Etienne G.J. Danchin | <p>The most devastating nematodes to worldwide agriculture are the root-knot nematodes with Meloidogyne incognita being the most widely distributed and damaging species. This parasitic and ecological success seem surprising given its supposed obli... | | Adaptation, Bioinformatics & Computational Biology, Evolutionary Ecology, Genome Evolution, Genotype-Phenotype, Molecular Evolution, Phylogenetics / Phylogenomics, Population Genetics / Genomics, Reproduction and Sex | Nicolas Galtier | 2018-08-24 09:02:33 | ||
17 May 2021

Relative time constraints improve molecular datingDating with constraintsRecommended by Cécile Ané based on reviews by David Duchêne and 1 anonymous reviewerEstimating the absolute age of diversification events is challenging, because molecular sequences provide timing information in units of substitutions, not years. Additionally, the rate of molecular evolution (in substitutions per year) can vary widely across lineages. Accurate dating of speciation events traditionally relies on non-molecular data. For very fast-evolving organisms such as SARS-CoV-2, for which samples are obtained over a time span, the collection times provide this external information from which we can learn the rate of molecular evolution and date past events (Boni et al. 2020). In groups for which the fossil record is abundant, state-of-the-art dating methods use fossil information to complement molecular data, either in the form of a prior distribution on node ages (Nguyen & Ho 2020), or as data modelled with a fossilization process (Heath et al. 2014). Dating is a challenge in groups that lack fossils or other geological evidence, such as very old lineages and microbial lineages. In these groups, horizontal gene transfer (HGT) events have been identified as informative about relative dates: the ancestor of the gene's donor must be older than the descendants of the gene's recipient. Previous work using HGTs to date phylogenies have used methodologies that are ad-hoc (Davín et al 2018) or employ a small number of HGTs only (Magnabosco et al. 2018, Wolfe & Fournier 2018). Szöllősi et al. (2021) present and validate a Bayesian approach to estimate the age of diversification events based on relative information on these ages, such as implied by HGTs. This approach is flexible because it is modular: constraints on relative node ages can be combined with absolute age information from fossil data, and with any substitution model of molecular evolution, including complex state-of-art models. To ease the computational burden, the authors also introduce a two-step approach, in which the complexity of estimating branch lengths in substitutions per site is decoupled from the complexity of timing the tree with branch lengths in years, accounting for uncertainty in the first step. Currently, one limitation is that the tree topology needs to be known, and another limitation is that constraints need to be certain. Users of this method should be mindful of the latter when hundreds of constraints are used, as done by Szöllősi et al. (2021) to date the trees of Cyanobacteria and Archaea. Szöllősi et al. (2021)'s method is implemented in RevBayes, a highly modular platform for phylogenetic inference, rapidly growing in popularity (Höhna et al. 2016). The RevBayes tutorial page features a step-by-step tutorial "Dating with Relative Constraints", which makes the method highly approachable. References: Boni MF, Lemey P, Jiang X, Lam TT-Y, Perry BW, Castoe TA, Rambaut A, Robertson DL (2020) Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nature Microbiology, 5, 1408–1417. https://doi.org/10.1038/s41564-020-0771-4 Davín AA, Tannier E, Williams TA, Boussau B, Daubin V, Szöllősi GJ (2018) Gene transfers can date the tree of life. Nature Ecology & Evolution, 2, 904–909. https://doi.org/10.1038/s41559-018-0525-3 Heath TA, Huelsenbeck JP, Stadler T (2014) The fossilized birth–death process for coherent calibration of divergence-time estimates. Proceedings of the National Academy of Sciences, 111, E2957–E2966. https://doi.org/10.1073/pnas.1319091111 Höhna S, Landis MJ, Heath TA, Boussau B, Lartillot N, Moore BR, Huelsenbeck JP, Ronquist F (2016) RevBayes: Bayesian Phylogenetic Inference Using Graphical Models and an Interactive Model-Specification Language. Systematic Biology, 65, 726–736. https://doi.org/10.1093/sysbio/syw021 Magnabosco C, Moore KR, Wolfe JM, Fournier GP (2018) Dating phototrophic microbial lineages with reticulate gene histories. Geobiology, 16, 179–189. https://doi.org/10.1111/gbi.12273 Nguyen JMT, Ho SYW (2020) Calibrations from the Fossil Record. In: The Molecular Evolutionary Clock: Theory and Practice (ed Ho SYW), pp. 117–133. Springer International Publishing, Cham. https://doi.org/10.1007/978-3-030-60181-2_8 Szollosi, G.J., Hoehna, S., Williams, T.A., Schrempf, D., Daubin, V., Boussau, B. (2021) Relative time constraints improve molecular dating. bioRxiv, 2020.10.17.343889, ver. 8 recommended and peer-reviewed by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.10.17.343889 Wolfe JM, Fournier GP (2018) Horizontal gene transfer constrains the timing of methanogen evolution. Nature Ecology & Evolution, 2, 897–903. https://doi.org/10.1038/s41559-018-0513-7 | Relative time constraints improve molecular dating | Gergely J Szollosi, Sebastian Hoehna, Tom A Williams, Dominik Schrempf, Vincent Daubin, Bastien Boussau | <p style="text-align: justify;">Dating the tree of life is central to understanding the evolution of life on Earth. Molecular clocks calibrated with fossils represent the state of the art for inferring the ages of major groups. Yet, other informat... | | Bioinformatics & Computational Biology, Genome Evolution, Phylogenetics / Phylogenomics | Cécile Ané | 2020-10-21 23:39:17 | ||
10 Jan 2020

Probabilities of tree topologies with temporal constraints and diversification shiftsFitting diversification models on undated or partially dated treesRecommended by Nicolas Lartillot based on reviews by Amaury Lambert, Dominik Schrempf and 1 anonymous reviewerPhylogenetic trees can be used to extract information about the process of diversification that has generated them. The most common approach to conduct this inference is to rely on a likelihood, defined here as the probability of generating a dated tree T given a diversification model (e.g. a birth-death model), and then use standard maximum likelihood. This idea has been explored extensively in the context of the so-called diversification studies, with many variants for the models and for the questions being asked (diversification rates shifting at certain time points or in the ancestors of particular subclades, trait-dependent diversification rates, etc). References [1] Didier, G. (2020) Probabilities of tree topologies with temporal constraints and diversification shifts. bioRxiv, 376756, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/376756 | Probabilities of tree topologies with temporal constraints and diversification shifts | Gilles Didier | <p>Dating the tree of life is a task far more complicated than only determining the evolutionary relationships between species. It is therefore of interest to develop approaches apt to deal with undated phylogenetic trees. The main result of this ... | | Bioinformatics & Computational Biology, Macroevolution | Nicolas Lartillot | 2019-01-30 11:28:58 | ||
23 Jan 2020

A novel workflow to improve multi-locus genotyping of wildlife species: an experimental set-up with a known model systemImproving the reliability of genotyping of multigene families in non-model organismsRecommended by François Rousset based on reviews by Sebastian Ernesto Ramos-Onsins, Helena Westerdahl and Thomas BigotThe reliability of published scientific papers has been the topic of much recent discussion, notably in the biomedical sciences [1]. Although small sample size is regularly pointed as one of the culprits, big data can also be a concern. The advent of high-throughput sequencing, and the processing of sequence data by opaque bioinformatics workflows, mean that sequences with often high error rates are produced, and that exact but slow analyses are not feasible. References [1] Ioannidis, J. P. A, Greenland, S., Hlatky, M. A., Khoury, M. J., Macleod, M. R., Moher, D., Schulz, K. F. and Tibshirani, R. (2014) Increasing value and reducing waste in research design, conduct, and analysis. The Lancet, 383, 166-175. doi: 10.1016/S0140-6736(13)62227-8 | A novel workflow to improve multi-locus genotyping of wildlife species: an experimental set-up with a known model system | Gillingham, Mark A. F., Montero, B. Karina, Wilhelm, Kerstin, Grudzus, Kara, Sommer, Simone and Santos, Pablo S. C. | <p>Genotyping novel complex multigene systems is particularly challenging in non-model organisms. Target primers frequently amplify simultaneously multiple loci leading to high PCR and sequencing artefacts such as chimeras and allele amplification... | | Bioinformatics & Computational Biology, Evolutionary Ecology, Genome Evolution, Molecular Evolution | François Rousset | Helena Westerdahl, Sebastian Ernesto Ramos-Onsins, Paul J. McMurdie , Arnaud Estoup, Vincent Segura, Jacek Radwan , Torbjørn Rognes , William Stutz , Kevin Vanneste , Thomas Bigot, Jill A. Hollenbach , Wieslaw Babik , Marie-Christin... | 2019-05-15 17:30:44 | |
12 Jul 2017

Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategiesTowards an integrated scenario to understand evolutionary patterns in A. thalianaRecommended by Xavier Picó based on reviews by Rafa Rubio de Casas and Xavier PicóNobody can ignore that a full understanding of evolution requires an integrated approach from both conceptual and methodological viewpoints. Although some life-history traits, e.g. flowering time, have long been receiving more attention than others, in many cases because the former are more workable than the latter, we must acknowledge that our comprehension about how evolution works is strongly biased and limited. In the Arabidopsis community, such an integration is making good progress as an increasing number of research groups worldwide are changing the way in which evolution is put to the test. This manuscript [1] is a good example of that as the authors raise an important issue in evolutionary biology by combining gene expression and flowering time data from different sources. In particular, the authors explore how variation in flowering time, which determines lifespan, and host immunity defenses co-vary, which is interpreted in terms of co-evolution between the two traits. Interestingly, the authors go beyond that pattern by separating lifespan-dependent from lifespan–independent defense genes, and by showing that defense genes with variants known to impact fitness in the field are among the genes whose expression co-varies most strongly with flowering time. Finally, these results are supported by a simple mathematical model indicating that such a relationship can also be expected theoretically. Overall, the readers will find many conceptual and methodological elements of interest in this manuscript. The idea that evolution is better understood under the scope of life history variation is really exciting and challenging, and in my opinion on the right track for disentangling the inherent complexities of evolutionary research. However, only when we face complexity, we also face its costs and burdens. In this particular case, the well-known co-variation between seed dormancy and flowering time is a missing piece, as well as the identification of (variation in) putative selective pressures accounting for the co-evolution between defense mechanisms and life history (seed dormancy vs. flowering time) along environmental gradients. More intellectual, technical and methodological challenges that with no doubt are totally worth it. Reference [1] Glander S, He F, Schmitz G, Witten A, Telschow A, de Meaux J. 2017. Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategies. bioRxiv ver.1 of June 19, 2017. doi: 10.1101/131136 | Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategies | Glander S, He F, Schmitz G, Witten A, Telschow A, de Meaux J | The selective impact of pathogen epidemics on host defenses can be strong but remains transient. By contrast, life-history shifts can durably and continuously modify the balance between costs and benefits of immunity, which arbitrates the evolutio... | | Adaptation, Evolutionary Ecology, Expression Studies, Life History, Phenotypic Plasticity, Quantitative Genetics, Species interactions | Xavier Picó | Sophie Karrenberg, Rafa Rubio de Casas, Xavier Picó | 2017-06-21 10:57:14 | |
18 Aug 2020
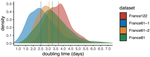
Early phylodynamics analysis of the COVID-19 epidemics in FranceSARS-Cov-2 genome sequence analysis suggests rapid spread followed by epidemic slowdown in FranceRecommended by B. Jesse Shapiro based on reviews by Luca Ferretti and 2 anonymous reviewersSequencing and analyzing SARS-Cov-2 genomes in nearly real time has the potential to quickly confirm (and inform) our knowledge of, and response to, the current pandemic [1,2]. In this manuscript [3], Danesh and colleagues use the earliest set of available SARS-Cov-2 genome sequences available from France to make inferences about the timing of the major epidemic wave, the duration of infections, and the efficacy of lockdown measures. Their phylodynamic estimates -- based on fitting genomic data to molecular clock and transmission models -- are reassuringly close to estimates based on 'traditional' epidemiological methods: the French epidemic likely began in mid-January or early February 2020, and spread relatively rapidly (doubling every 3-5 days), with people remaining infectious for a median of 5 days [4,5]. These transmission parameters are broadly in line with estimates from China [6,7], but are currently unknown in France (in the absence of contact tracing data). By estimating the temporal reproductive number (Rt), the authors detected a slowing down of the epidemic in the most recent period of the study, after mid-March, supporting the efficacy of lockdown measures. References [1] Grubaugh, N. D., Ladner, J. T., Lemey, P., Pybus, O. G., Rambaut, A., Holmes, E. C., & Andersen, K. G. (2019). Tracking virus outbreaks in the twenty-first century. Nature microbiology, 4(1), 10-19. doi: 10.1038/s41564-018-0296-2 | Early phylodynamics analysis of the COVID-19 epidemics in France | Gonché Danesh, Baptiste Elie,Yannis Michalakis, Mircea T. Sofonea, Antonin Bal, Sylvie Behillil, Grégory Destras, David Boutolleau, Sonia Burrel, Anne-Geneviève Marcelin, Jean-Christophe Plantier, Vincent Thibault, Etienne Simon-Loriere, Sylvie va... | <p>France was one of the first countries to be reached by the COVID-19 pandemic. Here, we analyse 196 SARS-Cov-2 genomes collected between Jan 24 and Mar 24 2020, and perform a phylodynamics analysis. In particular, we analyse the doubling time, r... | | Evolutionary Epidemiology, Molecular Evolution, Phylogenetics / Phylogenomics | B. Jesse Shapiro | 2020-06-04 13:13:57 | ||
11 Dec 2020

Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence dataPhylodynamics of hepatitis C virus reveals transmission dynamics within and between risk groups in LyonRecommended by David Rasmussen based on reviews by Chris Wymant and Louis DuPlessisGenomic epidemiology seeks to better understand the transmission dynamics of infectious pathogens using molecular sequence data. Phylodynamic methods have given genomic epidemiology new power to track the transmission dynamics of pathogens by combining phylogenetic analyses with epidemiological modeling. In recent year, applications of phylodynamics to chronic viral infections such as HIV and hepatitis C virus (HVC) have provided some of the best examples of how phylodynamic inference can provide valuable insights into transmission dynamics within and between different subpopulations or risk groups, allowing for more targeted interventions. References [1] Rasmussen, D. A., Volz, E. M., and Koelle, K. (2014). Phylodynamic inference for structured epidemiological models. PLoS Comput Biol, 10(4), e1003570. doi: https://doi.org/10.1371/journal.pcbi.1003570 | Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence data | Gonche Danesh, Victor Virlogeux, Christophe Ramière, Caroline Charre, Laurent Cotte, Samuel Alizon | <p>Opioid substitution and syringes exchange programs have drastically reduced hepatitis C virus (HCV) spread in France but HCV sexual transmission in men having sex with men (MSM) has recently arisen as a significant public health concern. The fa... | | Evolutionary Epidemiology, Phylogenetics / Phylogenomics | David Rasmussen | 2019-07-11 13:37:23 | ||
05 Feb 2019
The quiescent X, the replicative Y and the AutosomesReplication-independent mutations: a universal signature ?Recommended by Nicolas Galtier based on reviews by Marc Robinson-Rechavi and Robert LanfearMutations are the primary source of genetic variation, and there is an obvious interest in characterizing and understanding the processes by which they appear. One particularly important question is the relative abundance, and nature, of replication-dependent and replication-independent mutations - the former arise as cells replicate due to DNA polymerization errors, whereas the latter are unrelated to the cell cycle. A recent experimental study in fission yeast identified a signature of mutations in quiescent (=non-replicating) cells: the spectrum of such mutations is characterized by an enrichment in insertions and deletions (indels) compared to point mutations, and an enrichment of deletions compared to insertions [2]. References [1] Achaz, G., Gangloff, S., and Arcangioli, B. (2019). The quiescent X, the replicative Y and the Autosomes. BioRxiv, 351288, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/351288 | The quiescent X, the replicative Y and the Autosomes | Guillaume Achaz, Serge Gangloff, Benoit Arcangioli | <p>From the analysis of the mutation spectrum in the 2,504 sequenced human genomes from the 1000 genomes project (phase 3), we show that sexual chromosomes (X and Y) exhibit a different proportion of indel mutations than autosomes (A), ranking the... | | Bioinformatics & Computational Biology, Genome Evolution, Human Evolution, Molecular Evolution, Population Genetics / Genomics, Reproduction and Sex | Nicolas Galtier | 2018-07-25 10:37:48 |




