
Latest recommendations

| Id▲ | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
15 Dec 2016

POSTPRINT
Limiting opportunities for cheating stabilizes virulence in insect parasitic nematodesApplication of kin theory to long-standing problem in nematode production for biocontrolRecommended by Thomas Sappington and Ruth Arabelle HufbauerMuch research effort has been extended toward developing systems for managing soil inhabiting insect pests of crops with entomopathogenic nematodes as biocontrol agents. Although small plot or laboratory experiments may suggest a particular insect pest is vulnerable to management in this way, it is often difficult to scale-up nematode production for application at the field- and farm scale to make such a tactic viable. Part of the problem is that entomopathogenic nematode strains must be propagated by serial passage in vivo, because storage by freezing decreases fitness. At the same time, serial propagation results in loss of virulence (ability to infect) over generations in the laboratory, a phenomenon called attenuation. To probe the underlying reasons for development of attenuation, as a prerequisite to designing strategies to mitigate it, Shapiro-Ilan and Raymond [1] turned to evolutionary theory of social conflict as a possible explanatory framework. Virulence of entomopathogenic nematodes depends on a combination of virulence factors, like various proteases, secreted by both the nematode and symbiotic bacteria to overcome host defenses. Attenuation is characterized in part by a reduced production of these factors. Invasion of a host involves simultaneous attack by a group of nematodes ("cooperators"), which together neutralize host defenses enough to allow individuals to successfully invade. "Cheaters" in the invading population can avoid the metabolic costs of producing virulence factors while reaping the benefits of infecting the host made vulnerable by the cooperators in the population. The authors hypothesize that an increase in frequency of cheaters may contribute to attenuation of virulence during serial propagation in the laboratory. The evolutionary dynamics of cheater frequency in a population have been explored in many contexts as part of kin selection theory. Cheaters can increase in a population by outcompeting cooperators in a host if overall relatedness within the invading population is low. Conversely, frequency of altruism, or costly cooperation, increases in a population if relatedness is high, which is enhanced by low effective dispersal. However, a population that is too isolated can suffer from inbreeding effects, and competition will occur mainly among relatives, which decreases the fitness benefits of altruism. Shapiro-Ilan and Raymond [1] tested changes in virulence and reproductive output in a serially propagated entomopathogenic nematode, Heterorhabditis floridensis. They compared lines of high or low relatedness, manipulated via multiplicity of infection (MOI) rates (where a low dose of nematodes gives high relatedness and a high dose gives low relatedness); and under global or local competition, manipulated by pooling populations emerging from all or only two host cadavers per generation, respectively. As predicted, treatments of high relatedness (low MOI) and global competition had the greatest level of reproduction, while all lines of low relatedness (high MOI) evolved decreased reproduction and decreased virulence, which led to extinction. The key finding was that lines in the high relatedness (low MOI) and low (local) competition treatment exhibited the most stable virulence through the 12 generations tested. Thus, to minimize attenuation of virulence while maintaining fitness of recently isolated entomopathogenic nematodes, the authors recommend insect hosts be inoculated with low doses of nematodes from inocula pools from as few cadavers as possible. The application of evolutionary theory, with a clever experimental design, to an important problem in pest management makes this paper particularly noteworthy. Reference [1] Shapiro-Ilan D, Raymond B. 2016. Limiting opportunities for cheating stabilizes virulence in insect parasitic nematodes. Evolutionary Applications 9:462-470. doi: 10.1111/eva.12348 | Limiting opportunities for cheating stabilizes virulence in insect parasitic nematodes | Shapiro-Ilan D. and B. Raymond | Cooperative secretion of virulence factors by pathogens can lead to social conflict when cheating mutants exploit collective secretion, but do not contribute to it. If cheats outcompete cooperators within hosts, this can cause loss of virulence.... | | Adaptation, Behavior & Social Evolution, Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory, Experimental Evolution, Population Genetics / Genomics, Reproduction and Sex | Thomas Sappington | 2016-12-15 18:33:39 | ||
16 Dec 2016

POSTPRINT
Evolutionary robotics simulations help explain why reciprocity is rare in nature.Simulated robots and the evolution of reciprocityRecommended by Michael D Greenfield and Joël Meunier
Of the various forms of cooperative and altruistic behavior, reciprocity remains the most contentious. Humans certainly exhibit reciprocity – under certain circumstances – and various non-human animals behave in ways suggesting that they do as well. Thus, evolutionary biologists have sought to explain why non-relatives might engage in altruistic transactions when a substantial delay occurs between helping and compensation; i.e. an individual may be a donor today and a beneficiary tomorrow, or vice-versa. This quest, aided by game theory and computer modeling late in the past century, identified some strategies for reciprocal behavior that could work – in theory. But when biologists looked for confirmation of these strategies in animals they found little evidence that stood up to rigorous testing. In a recent paper André and Nolfi [1] offer a compelling reason for this observed rarity of reciprocity: Reciprocal behavior that animals might exhibit is a bit more complex than any of the game theoretic strategies, and even the simplest forms of realistic behavior would entail several nearly simultaneous mutations, an unlikely occurrence. André and Nolfi [1] relied on neural networks to test actors, robots that could evolve helping and reciprocal behavior from a basal level of selfishness. In an extensive series of simulations, they found that reciprocal behavior did not take hold in a population, largely because the various intermediates to full reciprocity were eliminated before the subsequent mutations occurred. The findings are satisfying given our current knowledge of animal behavior, but questions remain. Notably, how does one account for those rare cases in which reciprocity does meet all the criteria? The authors suggest some possibilities, but an analysis will await their next study. Reference [1] André J-B, Nolfi S. 2016. Evolutionary robotics simulations help explain why reciprocity is rare in nature. Scientific Reports 6:32785. doi: 10.1038/srep32785 | Evolutionary robotics simulations help explain why reciprocity is rare in nature. | André J-B, Nolfi S | The relative rarity of reciprocity in nature, contrary to theoretical predictions that it should be widespread, is currently one of the major puzzles in social evolution theory. Here we use evolutionary robotics to solve this puzzle. We show tha... | | Behavior & Social Evolution, Evolutionary Theory | Michael D Greenfield | 2016-12-16 18:08:31 | ||
17 Dec 2016
POSTPRINT
Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling studyPredicting HIV virulence evolution in response to widespread treatmentRecommended by Samuel Alizon and Roger Kouyos
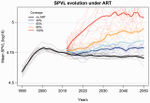
It is a classical result in the virulence evolution literature that treatments decreasing parasite replication within the host should select for higher replication rates, thus driving increased levels of virulence if the two are correlated. There is some evidence for this in vitro but very little in the field. HIV infections in humans offer a unique opportunity to go beyond the simple predictions that treatments should favour more virulent strains because many details of this host-parasite system are known, especially the link between set-point virus load, transmission rate and virulence. To tackle this question, Herbeck et al. [1] used a detailed individual-based model. This is original because it allows them to integrate existing knowledge from the epidemiology and evolution of HIV (e.g. recent estimates of the ‘heritability’ of set-point virus load from one infection to the next). This detailed model allows them to formulate predictions regarding the effect of different treatment policies; especially regarding the current policy switch away from treatment initiation based on CD4 counts towards universal treatment. The results show that, perhaps as expected from the theory, treatments based on the level of remaining host target cells (CD4 T cells) do not affect virulence evolution because they do not strongly affect the virulence level that maximizes HIV’s transmission potential. However, early treatments can lead to moderate increase in virulence within several years if coverage is high enough. These results seem quite robust to variation of all the parameters in realistic ranges. The great step forward in this model is the ability to obtain quantitative prediction regarding how a virus may evolve in response to public health policies. Here the main conclusion is that given our current knowledge in HIV biology, the risk of virulence evolution is perhaps more limited than expected from a direct application of virulence evolution model. Interestingly, the authors also conclude that recently observed increased in HIV virulence [2-3] cannot be explained by the impact of antiretroviral therapy alone; which raises the question about the main mechanism behind this increase. Finally, the authors make the interesting suggestion that “changing virulence is amenable to being monitored alongside transmitted drug resistance in sentinel surveillance”. References [1] Herbeck JT, Mittler JE, Gottlieb GS, Goodreau SM, Murphy JT, Cori A, Pickles M, Fraser C. 2016. Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling study. Virus Evolution 2:vew028. doi: 10.1093/ve/vew028 [2] Herbeck JT, Müller V, Maust BS, Ledergerber B, Torti C, et al. 2012. Is the virulence of HIV changing? A meta-analysis of trends in prognostic markers of HIV disease progression and transmission. AIDS 26:193-205. doi: 10.1097/QAD.0b013e32834db418 [3] Pantazis N, Porter K, Costagliola D, De Luca A, Ghosn J, et al. 2014. Temporal trends in prognostic markers of HIV-1 virulence and transmissibility: an observational cohort study. Lancet HIV 1:e119-26. doi: 10.1016/s2352-3018(14)00002-2 | Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling study | Herbeck JT, Mittler JE, Gottlieb GS, Goodreau SM, Murphy JT, Cori A, Pickles M, Fraser C | There are global increases in the use of HIV antiretroviral therapy (ART), guided by clinical benefits of early ART initiation and the efficacy of treatment as prevention of transmission. Separately, it has been shown theoretically and empirically... | | Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Epidemiology | Samuel Alizon | 2016-12-16 20:54:08 | ||
19 Dec 2016

POSTPRINT
Geographic body size variation in the periodical cicadas Magicicada: implications for life cycle divergence and local adaptationMegacicadas show a temperature-mediated converse Bergmann cline in body size (larger in the warmer south) but no body size difference between 13- and 17-year species pairsRecommended by Wolf Blanckenhorn and Thomas FlattPeriodical cicadas are a very prominent insect group in North America that are known for their large size, good looks, and loud sounds. However, they are probably known best to evolutionary ecologists because of their long juvenile periods of 13 or 17 years (prime numbers!), which they spend in the ground. Multiple related species living in the same area are often coordinated in emerging as adults during the same year, thereby presumably swamping any predators specialized on eating them. Reference [1] Koyama T, Ito H, Kakishima S, Yoshimura J, Cooley JR, Simon C, Sota T. 2015. Geographic body size variation in the periodical cicadas Magicicada: implications for life cycle divergence and local adaptation. Journal of Evolutionary Biology 28:1270-1277. doi: 10.1111/jeb.12653 | Geographic body size variation in the periodical cicadas Magicicada: implications for life cycle divergence and local adaptation | Koyama T, Ito H, Kakishima S, Yoshimura J, Cooley JR, Simon C, Sota T | Seven species in three species groups (Decim, Cassini and Decula) of periodical cicadas (*Magicicada*) occupy a wide latitudinal range in the eastern United States. To clarify how adult body size, a key trait affecting fitness, varies geographical... | | Adaptation, Evolutionary Ecology, Life History, Macroevolution, Phylogeography & Biogeography, Speciation | Wolf Blanckenhorn | 2016-12-19 10:39:22 | ||
20 Dec 2016

POSTPRINT
Experimental Evolution of Gene Expression and Plasticity in Alternative Selective RegimesGenetic adaptation counters phenotypic plasticity in experimental evolutionRecommended by Luis-Miguel Chevin and Stephanie BedhommeHow do phenotypic plasticity and adaptive evolution interact in a novel or changing environment? Does evolution by natural selection generally reinforce initially plastic phenotypic responses, or does it instead oppose them? And to what extent does evolution of a trait involve evolution of its plasticity? These questions have lied at the heart of research on phenotypic evolution in heterogeneous environments ever since it was realized that the environment is likely to affect the expression of many (perhaps most) characters of an individual. Importantly, this broad definition of phenotypic plasticity as change in the average phenotype of a given genotype in response to its environment of development (or expression) does not involve any statement about the adaptiveness of the plastic changes. Theory on the evolution of plasticity has devoted much effort to understanding how reaction norm should evolve under different regimes of environmental change in space and time, and depending on genetic constraints on reaction norm shapes. However on an empirically ground, the questions above have mostly been addressed for individual traits, often chosen a priori for their likeliness to exhibit adaptive plasticity, and we still lack more systematic answers. These can be provided by so-called ‘phenomic’ approaches, where a large number of traits are tracked without prior information on their biological or ecological function. A problem is that the number of phenotypic characters that can be measured in an organism is virtually infinite (and to some extent arbitrary), and that scaling issues makes it difficult to compare different sets of traits. Gene-expression levels offer a partial solution to this dilemma, as they can be considered as a very large number of traits (one per typed gene) that can be measured easily and uniformly (fold change in the number of reads in RNAseq). As for any traits, expression levels of different genes may be genetically correlated, to an extent that depends on their regulation mechanism: cis-regulatory sequences that only affect expression of neighboring genes are likely to cause independent gene expression, while more systematic modifiers of expression (e.g. trans-regulators such as transcription factors) may cause correlated genetic responses of the expression of many genes. Huang and Agrawal [1] have studied plasticity and evolution of gene expression level in young larvae of populations of Drosophila melanogaster that have evolved for about 130 generations under either a constant environment (salt or cadmium), or an environment that is heterogeneous in time or space (combining salt and cadmium). They report a wealth of results, of which we summarize the most striking here. First, among genes that (i) were initially highly plastic and (ii) evolved significant divergence in expression levels between constant environment treatments, the evolved divergence is predominantly in the opposite direction to the initial plastic response. This suggests that either plasticity was initially maladaptive, or the selective pressure changed during the evolutionary process (see below). This somewhat unexpected result strikingly mirrors that from a study published last year in Nature [2], where the same pattern was found for responses of guppies to the presence of predators. However, Huang and Agrawal [1] went beyond this study by deciphering the underlying mechanisms in several interesting ways. First, they showed that change in gene expression often occurred at genes close to SNPs with differentiated frequencies across treatments (but not at genes with differentiated SNPs in their coding sequences), suggesting that cis-regulatory sequences are involved. This is also suggested by the fact that changes in gene expression are mostly caused by the increased expression of only one allele at polymorphic loci, and is a first step towards investigating the genetic underpinnings of (co)variation in gene expression levels. Another interesting set of findings concerns evolution of plasticity in treatments with variable environments. To compare the gene-expression plasticity that evolved in these treatments to an expectation, the authors considered that the expression levels in populations maintained for a long time under constant salt or cadmium had reached an optimum. The differences between these expression levels were thus assumed to predict the level of plasticity that should evolve in a heterogeneous environment (with both cadmium and salt) under perfect environmental predictability. The authors showed that plasticity did evolve more in the expected direction in heterogeneous than in constant environments, resulting in better adapted final expression levels across environments. Taken collectively, these results provide an unprecedented set of patterns that are greatly informative on how plasticity and evolution interact in constant versus changing environments. But of course, interpretations in terms of adaptive versus maladaptive plasticity are more challenging, as the authors themselves admit. Even though environmentally determined gene expression is the basic mechanism underlying the phenotypic plasticity of most traits, it is extremely difficult to relate to more integrated phenotypes for which we can understand the selection pressures, especially in multicellular organisms. The authors have recently investigated evolutionary change of quantitative traits in these selected lines, so it might be possible to establish links between reaction norms for macroscopic traits to those for gene expression levels. Such an approach would also involve tracking gene expression throughout life, rather than only in young larvae as done here, thus putting phenotypic complexity back in the picture also for expression levels. Another difficulty is that a plastic response that was originally adaptive may be replaced by an opposite evolutionary response in the long run, without having to invoke initially maladaptive plasticity. For instance, the authors mention the possibility that a generic stress response is initially triggered by cadmium, but is eventually unnecessary and costly after evolution of genetic mechanisms for cadmium detoxification (a case of so-called genetic accommodation). In any case, this study by Huang and Agrawal [1], together with the one by Ghalambor et al. last year [2], reports novel and unexpected results, which are likely to stimulate researchers interested in plasticity and evolution in heterogeneous environments for the years to come. References [1] Huang Y, Agrawal AF. 2016. Experimental Evolution of Gene Expression and Plasticity in Alternative Selective Regimes. PLoS Genetics 12:e1006336. doi: 10.1371/journal.pgen.1006336 [2] Ghalambor CK, Hoke KL, Ruell EW, Fischer EK, Reznick DN, Hughes KA. 2015. Non-adaptive plasticity potentiates rapid adaptive evolution of gene expression in nature. Nature 525: 372-375. doi: 10.1038/nature15256 | Experimental Evolution of Gene Expression and Plasticity in Alternative Selective Regimes | Huang Y, Agrawal AF | Little is known of how gene expression and its plasticity evolves as populations adapt to different environmental regimes. Expression is expected to evolve adaptively in all populations but only those populations experiencing environmental heterog... | | Adaptation, Experimental Evolution, Expression Studies, Phenotypic Plasticity | Luis-Miguel Chevin | 2016-12-20 09:04:15 | ||
24 Jan 2017

POSTPRINT
Birth of a W sex chromosome by horizontal transfer of Wolbachia bacterial symbiont genomeA newly evolved W(olbachia) sex chromosome in pillbug!Recommended by Gabriel Marais and Sylvain CharlatIn some taxa such as fish and arthropods, closely related species can have different mechanisms of sex determination and in particular different sex chromosomes, which implies that new sex chromosomes are constantly evolving [1]. Several models have been developed to explain this pattern but empirical data are lacking and the causes of the fast sex chromosome turn over remain mysterious [2-4]. Leclerq et al. [5] in a paper that just came out in PNAS have focused on one possible explanation: Wolbachia. This widespread intracellular symbiont of arthropods can manipulate its host reproduction in a number of ways, often by biasing the allocation of resources toward females, the transmitting sex. Perhaps the most spectacular example is seen in pillbugs, where Wolbachia commonly turns infected males into females, thus doubling its effective transmission to grandchildren. Extensive investigations on this phenomenon were initiated 30 years ago in the host species Armadillidium vulgare. The recent paper by Leclerq et al. beautifully validates an hypothesis formulated in these pioneer studies [6], namely, that a nuclear insertion of the Wolbachia genome caused the emergence of new female determining chromosome, that is, a new sex chromosome. Many populations of A. vulgare are infected by the feminising Wolbachia strain wVulC, where the spread of the bacterium has also induced the loss of the ancestral female determining W chromosome (because feminized ZZ individuals produce females without transmitting any W). In these populations, all individuals carry two Z chromosomes, so that the bacterium is effectively the new sex-determining factor: specimens that received Wolbachia from their mother become females, while the occasional loss of Wolbachia from mothers to eggs allows the production of males. Intriguingly, studies from natural populations also report that some females are devoid both of Wolbachia and the ancestral W chromosome, suggesting the existence of new female determining nuclear factor, the hypothetical “f element”. Leclerq et al. [5] found the f element and decrypted its origin. By sequencing the genome of a strain carrying the putative f element, they found that a nearly complete wVulC genome got inserted in the nuclear genome and that the chromosome carrying the insertion has effectively become a new W chromosome. The insertion is indeed found only in females, PCRs and pedigree analysis tell. Although the Wolbachia-derived gene(s) that became sex-determining gene(s) remain to be identified among many possible candidates, the genomic and genetic evidence are clear that this Wolbachia insertion is determining sex in this pillbug strain. Leclerq et al. [5] also found that although this insertion is quite recent, many structural changes (rearrangements, duplications) have occurred compared to the wVulC genome, which study will probably help understand which bacterial gene(s) have retained a function in the nucleus of the pillbug. Also, in the future, it will be interesting to understand how and why exactly the nuclear inserted Wolbachia rose in frequency in the pillbug population and how the cytoplasmic Wolbachia was lost, and to tease apart the roles of selection and drift in this event. We highly recommend this paper, which provides clear evidence that Wolbachia has caused sex chromosome turn over in one species, opening the conjecture that it might have done so in many others. References [1] Bachtrog D, Mank JE, Peichel CL, Kirkpatrick M, Otto SP, Ashman TL, Hahn MW, Kitano J, Mayrose I, Ming R, Perrin N, Ross L, Valenzuela N, Vamosi JC. 2014. Tree of Sex Consortium. Sex determination: why so many ways of doing it? PLoS Biology 12: e1001899. doi: 10.1371/journal.pbio.1001899 [2] van Doorn GS, Kirkpatrick M. 2007. Turnover of sex chromosomes induced by sexual conflict. Nature 449: 909-912. doi: 10.1038/nature06178 [3] Cordaux R, Bouchon D, Grève P. 2011. The impact of endosymbionts on the evolution of host sex-determination mechanisms. Trends in Genetics 27: 332-341. doi: 10.1016/j.tig.2011.05.002 [4] Blaser O, Neuenschwander S, Perrin N. 2014. Sex-chromosome turnovers: the hot-potato model. American Naturalist 183: 140-146. doi: 10.1086/674026 [5] Leclercq S, Thézé J, Chebbi MA, Giraud I, Moumen B, Ernenwein L, Grève P, Gilbert C, Cordaux R. 2016. Birth of a W sex chromosome by horizontal transfer of Wolbachia bacterial symbiont genome. Proceeding of the National Academy of Science USA 113: 15036-15041. doi: 10.1073/pnas.1608979113 [6] Legrand JJ, Juchault P. 1984. Nouvelles données sur le déterminisme génétique et épigénétique de la monogénie chez le crustacé isopode terrestre Armadillidium vulgare Latr. Génétique Sélection Evolution 16: 57–84. doi: 10.1186/1297-9686-16-1-57 | Birth of a W sex chromosome by horizontal transfer of Wolbachia bacterial symbiont genome | Sébastien Leclercq, Julien Thézé, Mohamed Amine Chebbi, Isabelle Giraud, Bouziane Moumen, Lise Ernenwein, Pierre Grève, Clément Gilbert, and Richard Cordaux | Sex determination is an evolutionarily ancient, key developmental pathway governing sexual differentiation in animals. Sex determination systems are remarkably variable between species or groups of species, however, and the evolutionary forces und... | | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Reproduction and Sex, Species interactions | Gabriel Marais | 2017-01-13 15:15:51 | ||
03 Apr 2017
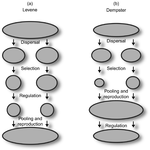
Things softly attained are long retained: Dissecting the Impacts of Selection Regimes on Polymorphism Maintenance in Experimental Spatially Heterogeneous EnvironmentsExperimental test of the conditions of maintenance of polymorphism under hard and soft selectionRecommended by Stephanie Bedhomme based on reviews by Joachim Hermisson and 2 anonymous reviewers
Theoretical work, initiated by Levene (1953) [1] and Dempster (1955) [2], suggests that within a given environment, the way populations are regulated and contribute to the next generation is a key factor for the maintenance of local adaptation polymorphism. In this theoretical context, hard selection describes the situation where the genetic composition of each population affects its contribution to the next generation whereas soft selection describes the case where the contribution of each population is fixed, whatever its genetic composition. Soft selection is able to maintain polymorphism, whereas hard selection invariably leads to the fixation of one of the alleles. Although the specific conditions (e.g. of migration between populations or drift level) in which this prediction holds have been studied in details by theoreticians, experimental tests have mainly failed, usually leading to the conclusion that the allele frequency dynamics was driven by other mechanisms in the experimental systems and conditions used. Gallet, Froissart and Ravigné [3] have set up a bacterial experimental system which allowed them to convincingly demonstrate that soft selection generates the conditions for polymorphism maintenance when hard selection does not, everything else being equal. The key ingredients of their experimental system are (1) the possibility to accurately produce hard and soft selection regimes when daily transferring the populations and (2) the ability to establish artificial well-characterized reproducible trade-offs. To do so, they used two genotypes resisting each one to one antibiotic and combined, across habitats, low antibiotic doses and difference in medium productivity. The experimental approach contains two complementary parts: the first one is looking at changes in the frequencies of two genotypes, initially introduced at around 50% each, over a small number of generations (ca 40) in different environments and selection regimes (soft/hard) and the second one is convincingly showing polymorphism protection by establishing that in soft selection regimes, the lowest fitness genotype is not eliminated even when introduced at low frequency. In this manuscript, a key point is the dialog between theoretical and experimental approaches. The experiments have been thought and designed to be as close as possible to the situations analysed in theoretical work. For example, the experimental polymorphism protection test (experiment 2) closely matches the equivalent analysis classically performed in theoretical approaches. This close fit between theory and experiment is clearly a strength of this study. This said, the experimental system allowing them to realise this close match also has some limitations. For example, changes in allele frequencies could only be monitored over a quite low number of generations because a longer time-scale would have allowed the contribution of de novo mutations and the likely emergence of a generalist genotype resisting to both antibiotics used to generate the local adaptation trade-offs. These limitations, as well as the actual significance of the experimental tests, are discussed in deep details in the manuscript. References [1] Levene H. 1953. Genetic equilibrium when more than one niche is available. American Naturalist 87: 331–333. doi: 10.1086/281792 [2] Dempster ER. 1955. Maintenance of genetic heterogeneity. Cold Spring Harbor Symposia on Quantitative Biology. 20: 25–32. doi: 10.1101/SQB.1955.020.01.005 [3] Gallet R, Froissart R, Ravigné V. 2017. Things softly attained are long retained: dissecting the impacts of selection regimes on polymorphism maintenance in experimental spatially heterogeneous environments. bioRxiv 100743; doi: 10.1101/100743 | Things softly attained are long retained: Dissecting the Impacts of Selection Regimes on Polymorphism Maintenance in Experimental Spatially Heterogeneous Environments | Romain Gallet, Rémy Froissart, Virginie Ravigné | <p>Predicting and managing contemporary adaption requires a proper understanding of the determinants of genetic variation. Spatial heterogeneity of the environment may stably maintain polymorphism when habitat contribution to the next generation c... | | Adaptation, Evolutionary Theory | Stephanie Bedhomme | 2017-01-17 11:06:21 | ||
18 Jan 2017

POSTPRINT
Associative Mechanisms Allow for Social Learning and Cultural Transmission of String Pulling in an InsectCulture in BumblebeesRecommended by Caroline Nieberding and Jacques J. M. van AlphenThis is an original paper [1] addressing the question whether cultural transmission occurs in insects and studying the mechanisms of such transmission. Often, culture-like phenomena require relatively sophisticated learning mechanisms, for example imitation and/or teaching. In insects, seemingly complex processes of social information acquisition, can sometimes instead be mediated by relatively simple learning mechanisms suggesting that cultural processes may not necessarily require sophisticated learning abilities. An important quality of this paper is to describe neatly the experimental protocols used for such typically complex behavioural analyses, providing a detailed understanding of the results while it remains a joy to read. This becomes rare in high impact journals. In a clever experimental design, individual bumblebees are trained to pull an artificial flower from under a Plexiglas table to get access to a reward, by pulling a string attached to the flower. Individuals that have learnt this task are then shown to inexperienced bees while performing this task. This results in a large proportion of the inexperienced observers learning to pull the string and getting access to the reward. Finally, the authors could then document the spread of the string pulling skill amongst other workers in the colony. Even when the originally trained individuals had died, the skill of string-pulling persisted in the colony, as long as they were challenged with the task. This shows that cultural transmission takes place within a colony. The authors provide evidence that the transmission of this behavior among individuals relies on a mix of social learning by local enhancement (bees were attracted to the location where they had observed a demonstrator) and of non-social, individual learning (pulling the string is learned by trial and errors and not by direct imitation of the conspecific). Data also show that simple associative mechanisms are enough and that stimulus enhancement was involved (bees were attracted to the string when its location was concordant with that during prior observation). The cleverly designed experiments use a paradigm (string-pulling) which has often been used to investigate cognitive abilities in vertebrates. Comparison with such studies indicate that bees, in some aspects of their learning, may not be different from birds, dogs, or apes as they also relied on the perceptual feedback provided by their actions, resulting in target movement to learn string pulling. The results of the study suggest that the combination of relatively simple forms of social learning and trial-and-error learning can mediate the acquisition of new skills and that bumblebees possess the essential cognitive elements for cultural transmission and in a broader sense, that the capacity of culture may be present within most animals. Can we expect behavioural innovation such as string pulling to occur in nature? Bombus terrestris colonies can reach a total of several hundreds foragers. In the experiments, foragers needed on average 5 rounds of observations with different demonstrators to learn how to pull the string. As individuals forage in a meadow full of flowers and conspecifics, transmission of behavioural innovations by repeated observations shouldn’t strike us as something impossible. Would the behavior survive through the winter? Bumblebee colonies are seasonal in northern areas and in the Mediterranean area but tropical species persists for several years. In seasonal species, all the workers die before winter and only new queens overwinter. So there is no possibility for seasonal foragers to transmit the technique overwinter. Only queens could potentially transmit it to new foragers in spring. However flowers are different in autumn and spring. Therefore, what queens have learnt about flowers in autumn would unlikely be useful in spring (providing that they can remember it). However there is no reason why the technique couldn't be transmitted from a colony to another between spring to autumn. Such transmission of new behaviour would more easily persist in perennial social insect colonies, like honeybees. Importantly, the bees used in these experiments came from a company whose rearing conditions are unknown, and only a few colonies were used for each experiment. As learning ability has a genetic basis [2-3], colonies differ in their ability to learn [4]. In this regard, the authors showed variation between individual bumblebees and between bumblebee colonies in learning ability. Hence, we would wish to know more about the level of genetic diversity in the wild, and of genetic differentiation between tested colonies (were they independent replicates?), to extrapolate the results to what may happen in the wild. Excitingly, the authors found 2 true innovators among the >400 individuals that were tested at least once for 5 min who would solve such a task without stepwise training or observation of skilled demonstrators, showing that behavioural innovation can occur in very small numbers of individuals, provided that an ecological trigger is provided (food reward). Hence this study shows that all ingredients for the long proposed “social heredity” theory proposed by Baldwin in 1896 are available in this organism, suggesting that social transmission of behavioural innovations could technically act as an additional mechanism for adaptive evolution [5], next to genetic evolution that may take longer to produce adaptive evolution. The question remains whether the behavioural innovations are arising from standing genetic variation in the bees, or do not need a firm genetic background to appear. References [1] Alem S, Perry CJ, Zhu X, Loukola OJ, Ingraham T, Søvik E, Chittka L. 2016. Associative mechanisms allow for social learning and cultural transmission of string pulling in an insect. PloS Biology 14:e1002564. doi: 10.1371/journal.pbio.1002564 [2] Mery F, Kawecki TJ. 2002. Experimental evolution of learning ability in fruit flies. Proceeding of the National Academy of Science USA 99:14274-14279. doi: 10.1073/pnas.222371199 [3] Mery F, Belay AT, So AKC, Sokolowski MB, Kawecki TJ. 2007. Natural polymorphism affecting learning and memory in Drosophila. Proceeding of the National Academy of Science USA 104:13051-13055. doi: 10.1073/pnas.0702923104 [4] Raine NE, Chittka L. 2008. The correlation of learning speed and natural foraging success in bumble-bees. Proceeding of the Royal Society of London 275: 803-808. doi : 10.1098/rspb.2007.1652 [5] Baldwin JM. 1896. A New Factor in Evolution. American Naturalist 30:441-451 and 536-553. doi: 10.1086/276408 | Associative Mechanisms Allow for Social Learning and Cultural Transmission of String Pulling in an Insect | Alem S, Perry CJ, Zhu X, Loukola OJ, Ingraham T, Søvik E, Chittka L | Social insects make elaborate use of simple mechanisms to achieve seemingly complex behavior and may thus provide a unique resource to discover the basic cognitive elements required for culture, i.e., group-specific behaviors that spread from “inn... | | Behavior & Social Evolution, Evolutionary Ecology, Non Genetic Inheritance, Phenotypic Plasticity | Caroline Nieberding | 2017-01-18 10:49:03 | ||
26 Sep 2017

Lacking conservation genomics in the giant Galápagos tortoiseA genomic perspective is needed for the re-evaluation of species boundaries, evolutionary trajectories and conservation strategies for the Galápagos giant tortoisesRecommended by Michael C. Fontaine based on reviews by 4 anonymous reviewersGenome-wide data obtained from even a small number of individuals can provide unprecedented levels of detail about the evolutionary history of populations and species [1], determinants of genetic diversity [2], species boundaries and the process of speciation itself [3]. Loire and Galtier [4] present a clear example, using the emblematic Galápagos giant tortoise (Chelonoidis nigra), of how multi-species comparative population genomic approaches can provide valuable insights about population structure and species delimitation even when sample sizes are limited but the number of loci is large and distributed across the genome. Galápagos giant tortoises are endemic to the Galápagos Islands and are currently recognized as an endangered, multi-species complex including both extant and extinct taxa. Taxonomic definitions are based on morphology, geographic isolation and population genetic evidence based on short DNA sequences of the mitochondrial genome (mtDNA) and/or a dozen or so nuclear microsatellite loci [5-8]. The species complex enjoys maximal protection. Population recoveries have been quite successful and spectacular conservation programs based on mitochondrial genes and microsatellites are ongoing. This includes for example individual translocations, breeding program, “hybrid” sterilization or removal, and resurrection of extinct lineages). In 2013, Loire et al. [9] provided the first population genomic analyses based on genome scale data (~1000 coding loci derived from blood-transcriptomes) from five individuals, encompassing three putative “species”: Chelonnoidis becki, C. porteri and C. vandenburghi. Their results raised doubts about the validity/accuracy of the currently accepted designations of “genetic distinctiveness”. However, the implications for conservation and management have remained unnoticed. In 2017, Loire and Galtier [4] have re-appraised this issue using an original multi-species comparative population genomic analysis of their previous data set [9]. Based on a comparison of 53 animal species, they show that both the level of genome-wide neutral diversity (πS) and level of population structure estimated using the inbreeding coefficient (F) are much lower than would be expected from a sample covering multiple species. The observed values are more comparable to those typically reported at the “among population” level within a single species such as human (Homo sapiens). The authors go to great length to assess the sensitivity of their method to detect population structure (or lack thereof) and show that their results are robust to potential issues, such as contamination and sequencing errors that can occur with Next Generation Sequencing techniques; and biases related to the small sample size and sub-sampling of individuals. They conclude that published mtDNA and microsatellite-based assessment of population structure and species designations may be biased towards over-splitting. This manuscript is a very good read as it shows the potential of the now relatively affordable genome-wide data for helping to both resolve and clarify population and species boundaries, illuminate demographic trends, evolutionary trajectories of isolated groups, patterns of connectivity among them, and test for evidence of local adaptation and even reproductive isolation. The comprehensive information provided by genome-wide data can critically inform and assist the development of the best strategies to preserve endangered populations and species. Loire and Galtier [4] make a strong case for applying genomic data to the Galápagos giant tortoises, which is likely to redirect conservation efforts more effectively and at lower cost. The case of the Galápagos giant tortoises is certainly a very emblematic example, which will find an echo in many other endangered species conservation programs. References [1] Li H and Durbin R. 2011. Inference of human population history from individual whole-genome sequences. Nature, 475: 493–496. doi: 10.1038/nature10231 [2] Romiguier J, Gayral P, Ballenghien M, Bernard A, Cahais V, Chenuil A, Chiari Y, Dernat R, Duret L, Faivre N, Loire E, Lourenco JM, Nabholz B, Roux C, Tsagkogeorga G, Weber AA-T, Weinert LA, Belkhir K, Bierne N, Glémin S and Galtier N. 2014. Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature, 515: 261–263. doi: 10.1038/nature13685 [3] Roux C, Fraïsse C, Romiguier J, Anciaux Y, Galtier N and Bierne N. 2016. Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS Biology, 14: e2000234. doi: 10.1371/journal.pbio.2000234 [4] Loire E and Galtier N. 2017. Lacking conservation genomics in the giant Galápagos tortoise. bioRxiv 101980, ver. 4 of September 26, 2017. doi: 10.1101/101980 [5] Beheregaray LB, Ciofi C, Caccone A, Gibbs JP and Powell JR. 2003. Genetic divergence, phylogeography and conservation units of giant tortoises from Santa Cruz and Pinzón, Galápagos Islands. Conservation Genetics, 4: 31–46. doi: 10.1023/A:1021864214375 [6] Ciofi C, Milinkovitch MC, Gibbs JP, Caccone A and Powell JR. 2002. Microsatellite analysis of genetic divergence among populations of giant Galápagos tortoises. Molecular Ecology, 11: 2265–2283. doi: 10.1046/j.1365-294X.2002.01617.x [7] Garrick RC, Kajdacsi B, Russello MA, Benavides E, Hyseni C, Gibbs JP, Tapia W and Caccone A. 2015. Naturally rare versus newly rare: demographic inferences on two timescales inform conservation of Galápagos giant tortoises. Ecology and Evolution, 5: 676–694. doi: 10.1002/ece3.1388 [8] Poulakakis N, Edwards DL, Chiari Y, Garrick RC, Russello MA, Benavides E, Watkins-Colwell GJ, Glaberman S, Tapia W, Gibbs JP, Cayot LJ and Caccone A. 2015. Description of a new Galápagos giant tortoise species (Chelonoidis; Testudines: Testudinidae) from Cerro Fatal on Santa Cruz island. PLoS ONE, 10: e0138779. doi: 10.1371/journal.pone.0138779 [9] Loire E, Chiari Y, Bernard A, Cahais V, Romiguier J, Nabholz B, Lourenço JM and Galtier N. 2013. Population genomics of the endangered giant Galápagos tortoise. Genome Biology, 14: R136. doi: 10.1186/gb-2013-14-12-r136 | Lacking conservation genomics in the giant Galápagos tortoise | Etienne Loire, Nicolas Galtier | <p>Conservation policy in the giant Galápagos tortoise, an iconic endangered animal, has been assisted by genetic markers for ~15 years: a dozen loci have been used to delineate thirteen (sub)species, between which hybridization is prevented. Here... | | Evolutionary Applications, Population Genetics / Genomics, Speciation, Systematics / Taxonomy | Michael C. Fontaine | 2017-01-21 15:34:00 | ||
22 May 2017
Can Ebola Virus evolve to be less virulent in humans?A new hypothesis to explain Ebola's high virulenceRecommended by Virginie Ravigné and François Blanquart based on reviews by Virginie Ravigné and François Blanquart
The tragic 2014-2016 Ebola outbreak that resulted in more than 28,000 cases and 11,000 deaths in West Africa [1] has been a surprise to the scientific community. Before 2013, the Ebola virus (EBOV) was known to produce recurrent outbreaks in remote villages near tropical rainforests in Central Africa, never exceeding a few hundred cases with very high virulence. Both EBOV’s ability to circulate for several months in large urban human populations and its important mutation rate suggest that EBOV’s virulence could evolve and to some extent adapt to human hosts [2]. Up to now, the high virulence of EBOV in humans was generally thought to be maladaptive, the virus being adapted to circulating in wild animal populations (e.g. fruit bats [3]). As a logical consequence, EBOV virulence could be expected to decrease during long epidemics in humans. The present paper by Sofonea et al. [4] challenges this view and explores how, given EBOV’s life cycle and known epidemiological parameters, virulence is expected to evolve in the human host during long epidemics. The main finding of the paper is that there is no chance that EBOV’s virulence decreases in the short and long terms. The main underlying mechanism is that EBOV is also transmitted by dead bodies, which limits the cost of virulence. In itself the idea that selection should select for higher virulence in diseases that are also transmitted after host death will sound intuitive for most evolutionary epidemiologists. The accomplishment of the paper is to make a very strong case that the parameter range where virulence could decrease is very small. The paper further provides scientifically grounded arguments in favor of the safe management of corpses. Safe burial of corpses is culturally difficult to impose. The present paper shows that in addition to instantaneously decreasing the spread of the virus, safe burial may limit virulence increase in the short term and favor of less virulent strains in the long term. Altogether these results make a timely and important contribution to the knowledge and understanding of EBOV. References [1] World Health Organization. 2016. WHO: Ebola situation report - 10 June 2016. [2] Kupferschmidt K. 2014. Imagining Ebola’s next move. Science 346: 151–152. doi: 10.1126/science.346.6206.151 [3] Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, Délicat A, Paweska, Gonzalez JP and Swanepoel R. 2005. Fruit bats as reservoirs of Ebola virus. Nature 438: 575–576. doi: 10.1038/438575a [4] Sofonea MT, Aldakak L, Boullosa LFVV and Alizon S. 2017. Can Ebola Virus evolve to be less virulent in humans? bioRxiv 108589, ver. 3 of 19th May 2017; doi: 10.1101/108589 | Can Ebola Virus evolve to be less virulent in humans? | Mircea T. Sofonea, Lafi Aldakak, Luis Fernando Boullosa, Samuel Alizon | Understanding Ebola Virus (EBOV) virulence evolution is not only timely but also raises specific questions because it causes one pf the most virulent human infections and it is capable of transmission after the death of its host. Using a compartme... | | Evolutionary Epidemiology | Virginie Ravigné | 2017-02-15 13:25:58 |




