
Latest recommendations

| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender▲ | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
02 Jan 2019
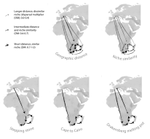
Leaps and bounds: geographical and ecological distance constrained the colonisation of the Afrotemperate by EricaThe colonization history of largely isolated habitatsRecommended by Andrea S. Meseguer based on reviews by Simon Joly, Florian Boucher and 2 anonymous reviewersThe build-up of biodiversity is the result of in situ speciation and immigration, with the interplay between geographical distance and ecological suitability determining the probability of an organism to establish in a new area. The relative contribution of these factors have long interested biogeographers, in particular to explain the distribution of organisms adapted to habitats that remained largely isolated, such as the colonization of oceanic islands or land waters. The focus of this study is the formation of the afrotemperate flora; patches of temperate vegetation separated by thousands of kilometers in Africa, with high levels of endemism described in the Cape region, the Drakensberg range and the high mountains of tropical east Africa [1]. The floristic affinities between these centers of endemism have frequently been explored but the origin of many afrotemperate lineages remains enigmatic [2]. References [1] Linder, H.P. 1990. On the relationship between the vegetation and floras of the Afromontane and the Cape regions of Africa. Mitteilungen aus dem Institut für Allgemeine Botanik Hamburg 23b:777–790. | Leaps and bounds: geographical and ecological distance constrained the colonisation of the Afrotemperate by Erica | Michael D. Pirie, Martha Kandziora, Nicolai M. Nuerk, Nicholas C. Le Maitre, Ana Laura Mugrabi de Kuppler, Berit Gehrke, Edward G.H. Oliver, and Dirk U. Bellstedt | <p>The coincidence of long distance dispersal and biome shift is assumed to be the result of a multifaceted interplay between geographical distance and ecological suitability of source and sink areas. Here, we test the influence of these factors o... | | Phylogeography & Biogeography | Andrea S. Meseguer | 2018-04-09 10:10:04 | ||
20 Nov 2017
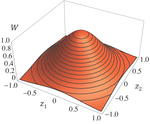
Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selectionUnderstanding genetic variance, load, and inbreeding depression with selfingRecommended by Aneil F. Agrawal based on reviews by Frédéric Guillaume and 1 anonymous reviewerA classic problem in evolutionary biology is to understand the genetic variance in fitness. The simplest hypothesis is that variation exists, even in well-adapted populations, as a result of the balance between mutational input and selective elimination. This variation causes a reduction in mean fitness, known as the mutation load. Though mutation load is difficult to quantify empirically, indirect evidence of segregating genetic variation in fitness is often readily obtained by comparing the fitness of inbred and outbred offspring, i.e., by measuring inbreeding depression. Mutation-selection balance models have been studied as a means of understanding the genetic variance in fitness, mutation load, and inbreeding depression. Since their inception, such models have increased in sophistication, allowing us to ask these questions under more realistic and varied scenarios. The new theoretical work by Abu Awad and Roze [1] is a substantial step forward in understanding how arbitrary levels of self-fertilization affect variation, load and inbreeding depression under mutation-selection balance. References [1] Abu Awad D and Roze D. 2017. Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selection. bioRxiv, 180000, ver. 4 of 17th November 2017. doi: 10.1101/180000 [2] Lande R. 1977. The influence of the mating system on the maintenance of genetic variability in polygenic characters. Genetics 86: 485–498. [3] Charlesworth D and Charlesworth B. 1987. Inbreeding depression and its evolutionary consequences. Annual Review of Ecology and Systematics. 18: 237–268. doi: 10.1111/10.1146/annurev.es.18.110187.001321 [4] Lande R and Porcher E. 2015. Maintenance of quantitative genetic variance under partial self-fertilization, with implications for the evolution of selfing. Genetics 200: 891–906. doi: 10.1534/genetics.115.176693 [5] Roze D. 2015. Effects of interference between selected loci on the mutation load, inbreeding depression, and heterosis. Genetics 201: 745–757. doi: 10.1534/genetics.115.178533 [6] Martin G and Lenormand T. 2006. A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species. Evolution 60: 893–907. doi: 10.1111/j.0014-3820.2006.tb01169.x [7] Martin G, Elena SF and Lenormand T. 2007. Distributions of epistasis in microbes fit predictions from a fitness landscape model. Nature Genetics 39: 555–560. doi: 10.1038/ng1998 | Effects of partial selfing on the equilibrium genetic variance, mutation load and inbreeding depression under stabilizing selection | Diala Abu Awad and Denis Roze | The mating system of a species is expected to have important effects on its genetic diversity. In this paper, we explore the effects of partial selfing on the equilibrium genetic variance Vg, mutation load L and inbreeding depression δ under stabi... | | Evolutionary Theory, Population Genetics / Genomics, Quantitative Genetics, Reproduction and Sex | Aneil F. Agrawal | 2017-08-26 09:29:20 | ||
14 Mar 2017

POSTPRINT
Evolution of multiple sensory systems drives novel egg-laying behavior in the fruit pest Drosophila suzukiiA valuable work lying at the crossroad of neuro-ethology, evolution and ecology in the fruit pest Drosophila suzukiiRecommended by Arnaud Estoup and Ruth Arabelle HufbauerAdaptations to a new ecological niche allow species to access new resources and circumvent competitors and are hence obvious pathways of evolutionary success. The evolution of agricultural pest species represents an important case to study how a species adapts, on various timescales, to a novel ecological niche. Among the numerous insects that are agricultural pests, the ability to lay eggs (or oviposit) in ripe fruit appears to be a recurrent scenario. Fruit flies (family Tephritidae) employ this strategy, and include amongst their members some of the most destructive pests (e.g., the olive fruit fly Bactrocera olea or the medfly Ceratitis capitata). In their ms, Karageorgi et al. [1] studied how Drosophila suzukii, a new major agricultural pest species that recently invaded Europe and North America, evolved the novel behavior of laying eggs into undamaged fresh fruit. The close relatives of D. suzukii lay their eggs on decaying plant substrates, and thus this represents a marked change in host use that links to substantial economic losses to the fruit industry. Although a handful of studies have identified genetic changes causing new behaviors in various species, the question of the evolution of behavior remains a largely uncharted territory. The study by Karageorgi et al. [1] represents an original and most welcome contribution in this domain for a non-model species. Using clever behavioral experiments to compare D. suzukii to several related Drosophila species, and complementing those results with neurogenetics and mutant analyses using D. suzukii, the authors nicely dissect the sensory changes at the origin of the new egg-laying behavior. The experiments they describe are easy to follow, richly illustrate through figures and images, and particularly well designed to progressively decipher the sensory bases driving oviposition of D. suzukii on ripe fruit. Altogether, Karageorgi et al.’s [1] results show that the egg-laying substrate preference of D. suzukii has considerably evolved in concert with its morphology (especially its enlarged, serrated ovipositor that enables females to pierce the skin of many ripe fruits). Their observations clearly support the view that the evolution of traits that make D. suzukii an agricultural pest included the modification of several sensory systems (i.e. mechanosensation, gustation and olfaction). These differences between D. suzukii and its close relatives collectively underlie a radical change in oviposition behavior, and were presumably instrumental in the expansion of the ecological niche of the species. The authors tentatively propose a multi-step evolutionary scenario from their results with the emergence of D. suzukii as a pest species as final outcome. Such formalization represents an interesting evolutionary model-framework that obviously would rely upon further data and experiments to confirm and refine some of the evolutionary steps proposed, especially the final and recent transition of D. suzukii from non-invasive to invasive species. References [1] Karageorgi M, Bräcker LB, Lebreton S, Minervino C, Cavey M, Siju KP, Grunwald Kadow IC, Gompel N, Prud’homme B. 2017. Evolution of multiple sensory systems drives novel egg-laying behavior in the fruit pest Drosophila suzukii. Current Biology, 27: 1-7. doi: 10.1016/j.cub.2017.01.055 | Evolution of multiple sensory systems drives novel egg-laying behavior in the fruit pest Drosophila suzukii | Marianthi Karageorgi, Lasse B. Bräcker, Sébastien Lebreton, Caroline Minervino, Matthieu Cavey, K.P. Siju, Ilona C. Grunwald Kadow, Nicolas Gompel, Benjamin Prud’homme | The rise of a pest species represents a unique opportunity to address how species evolve new behaviors and adapt to novel ecological niches. We address this question by studying the egg-laying behavior of Drosophila suzukii, an invasive agricultur... | | Adaptation, Behavior & Social Evolution, Evo-Devo, Evolutionary Applications, Evolutionary Ecology, Expression Studies, Genotype-Phenotype, Macroevolution, Molecular Evolution | Arnaud Estoup | 2017-03-13 17:42:00 | ||
03 Oct 2018
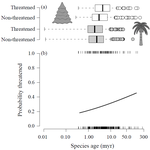
Range size dynamics can explain why evolutionarily age and diversification rate correlate with contemporary extinction risk in plantsAre both very young and the very old plant lineages at heightened risk of extinction?Recommended by Arne Mooers based on reviews by Dan Greenberg and 1 anonymous reviewerHuman economic activity is responsible for the vast majority of ongoing extinction, but that does not mean lineages are being affected willy-nilly. For amphibians [1] and South African flowering plants [2], young species have a somewhat higher than expected chance of being threatened with extinction. In contrast, older Australian marsupial lineages seem to be more at risk [3]. Both of the former studies suggested that situations leading to peripheral isolation might simultaneously increase ongoing speciation and current threat via small geographic range, while the authors of the latter study suggested that older species might have evolved increasingly narrow niches. Here, Andrew Tanentzap and colleagues [4] dig deeper into the putative links between species age, niche breadth and threat status. Across 500-some plant genera worldwide, they find that, indeed, ""younger"" species (i.e. from younger and faster-diversifying genera) were more likely to be listed as imperiled by the IUCN, consistent with patterns for amphibians and African plants. Given this, results from their finer-level analyses of conifers are initially bemusing: here, ""older"" (i.e., on longer terminal branches) species were at higher risk. This would make conifers more like Australian marsupials, with the rest of the plants being more like amphibians. However, here where the data were more finely grained, the authors detected a second interesting pattern: using an intriguing matched-pair design, they detect a signal of conifer species niches seemingly shrinking as a function of age. The authors interpret this as consistent with increasing specialization, or loss of ancestral warm wet habitat, over paleontological time. It is true that conifers in general are older than plants more generally, with some species on branches that extend back many 10s of millions of years, and so a general loss of suitable habitat makes some sense. If so, both the pattern for all plants (small initial ranges heightening extinction) and the pattern for conifers (eventual increasing specialization or habitat contraction heightening extinction) could occur, each on a different time scale. As a coda, the authors detected no effect of age on threat status in palms; however, this may be both because palms have already lost species to climate-change induced extinction, and because they are thought to speciate more via long-distance dispersal and adaptive divergence then via peripheral isolation. References [1] Greenberg, D. A., & Mooers, A. Ø. (2017). Linking speciation to extinction: Diversification raises contemporary extinction risk in amphibians. Evolution Letters, 1, 40–48. doi: 10.1002/evl3.4 | Range size dynamics can explain why evolutionarily age and diversification rate correlate with contemporary extinction risk in plants | Andrew J. Tanentzap, Javier Igea, Matthew G. Johnston, Matthew J. Larcombe | <p>Extinction threatens many species, yet few factors predict this risk across the plant Tree of Life (ToL). Taxon age is one factor that may associate with extinction if occupancy of geographic and adaptive zones varies with time, but evidence fo... | | Macroevolution, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Arne Mooers | 2018-02-01 21:01:19 | ||
14 Dec 2016
POSTPRINT
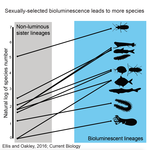
High Rates of Species Accumulation in Animals with Bioluminescent Courtship DisplaysBioluminescent sexually selected traits as an engine for biodiversity across animal speciesRecommended by Astrid Groot and Carole SmadjaIn evolutionary biology, sexual selection is hypothesized to increase speciation rates in animals, as theory predicts that sexual selection will contribute to phenotypic diversification and affect rates of species accumulation at macro-evolutionary time scales. However, testing this hypothesis and gathering convincing evidence have proven difficult. Although some studies have shown a strong correlation between proxies of sexual selection and species diversity (mostly in birds), this relationship relies on some assumptions on the link between these proxies and the strength of sexual selection and is not detected in some other taxa, making taxonomically widespread conclusions impossible. In a recent study published in Current Biology [1], Ellis and Oakley provide strong evidence that bioluminescent sexual displays have driven high species richness in taxonomically diverse animal lineages, providing a crucial link between sexual selection and speciation. Ellis and Oakley [1] explored the scientific literature for well-resolved evolutionary trees with branches containing bioluminescent lineages and identified lineages that use light for courtship or camouflage in a wide range of marine and terrestrial taxa including insects, crustaceans, cephalopods, segmented worms, and fishes. The researchers counted the number of species in each bioluminescent clade and found that all groups with light-courtship displays had more species and faster rates of species accumulation than their non-luminous most closely related sister lineages or ancestors. In contrast, those groups that used bioluminescence for predator avoidance had a lower than expected rate of species richness on average. Nicely encompassing a diversity of taxa and neatly controlling for the rate of species accumulation of the encompassing clade, the results of Ellis and Oakley are clear-cut and provide the most comprehensive evidence to date for the hypothesis that sexual displays can act as drivers of speciation. One question this study incites is what is happening in terms of sexual selection in species displaying defensive bioluminescence or no bioluminescence at all: do those lineages use no mating signals at all or other mating signals that are less apparent, and will those experience lower levels of sexual selection than bioluminescent mating signals, i.e. consistent with Ellis and Oakley results? It would also be interesting to investigate the diversification rates in animal species using other modalities, such as chemical, acoustic or any other type of signals used by males, females or both sexes, to determine what types of sexual signals may be more generally drivers of speciation. References [1] Ellis EA, Oakley TH. 2016. High Rates of Species Accumulation in Animals with Bioluminescent Courtship Displays. Current Biology 26:1916–1921. doi: 10.1016/j.cub.2016.05.043 [2] Davis MP, Holcroft NI, Wiley EO, Sparks JS, Smith WL. 2014. Species-specific bioluminescence facilitates speciation in the deep sea. Marine Biology 161:11391148. doi: 10.1007/s00227-014-2406-x [3] Davis MP, Sparks JS, Smith WL. 2016. Repeated and Widespread Evolution of Bioluminescence in Marine Fishes. PLoS One 11:e0155154. doi: 10.1371/journal.pone.0155154 [4] Claes JM, Nilsson D-E, Mallefet J, Straube N. 2015. The presence of lateral photophores correlates with increased speciation in deep-sea bioluminescent sharks. Royal Society Open Science 2:150219. doi: 10.1098/rsos.150219 | High Rates of Species Accumulation in Animals with Bioluminescent Courtship Displays | Ellis EA, Oakley TH | One of the great mysteries of evolutionary biology is why closely related lineages accumulate species at different rates. Theory predicts that populations undergoing strong sexual selection will more quickly differentiate because of increased pote... | | Adaptation, Evolutionary Ecology, Sexual Selection, Speciation | Astrid Groot | 2016-12-14 19:01:59 | ||
25 Jun 2020

Transcriptional differences between the two host strains of Spodoptera frugiperda (Lepidoptera: Noctuidae)Speciation through selection on mitochondrial genes?Recommended by Astrid Groot based on reviews by Heiko Vogel and Sabine HaennigerWhether speciation through ecological specialization occurs has been a thriving research area ever since Mayr (1942) stated this to play a central role. In herbivorous insects, ecological specialization is most likely to happen through host plant differentiation (Funk et al. 2002). Therefore, after Dorothy Pashley had identified two host strains in the Fall armyworm (FAW), Spodoptera frugiperda, in 1988 (Pashley 1988), researchers have been trying to decipher the evolutionary history of these strains, as this seems to be a model species in which speciation is currently occurring through host plant specialization. Even though FAW is a generalist, feeding on many different host plant species (Pogue 2002) and a devastating pest in many crops, Pashley identified a so-called corn strain and a so-called rice strain in Puerto Rico. Genetically, these strains were found to differ mostly in an esterase, although later studies showed additional genetic differences and markers, mostly in the mitochondrial COI and the nuclear TPI. Recent genomic studies showed that the two strains are overall so genetically different (2% of their genome being different) that these two strains could better be called different species (Kergoat et al. 2012). So far, the most consistent differences between the strains have been their timing of mating activities at night (Schoefl et al. 2009, 2011; Haenniger et al. 2019) and hybrid incompatibilities (Dumas et al. 2015; Kost et al. 2016). Whether and to what extent host plant preference or performance contributed to the differentiation of these sympatrically occurring strains has remained unclear. References [1] Dumas, P. et al. (2015). Spodoptera frugiperda (Lepidoptera: Noctuidae) host-plant variants: two host strains or two distinct species?. Genetica, 143(3), 305-316. doi: 10.1007/s10709-015-9829-2 | Transcriptional differences between the two host strains of Spodoptera frugiperda (Lepidoptera: Noctuidae) | Marion Orsucci, Yves Moné, Philippe Audiot, Sylvie Gimenez, Sandra Nhim, Rima Naït-Saïdi, Marie Frayssinet, Guillaume Dumont, Jean-Paul Boudon, Marin Vabre, Stéphanie Rialle, Rachid Koual, Gael J. Kergoat, Rodney N. Nagoshi, Robert L. Meagher, Emm... | <p>Spodoptera frugiperda, the fall armyworm (FAW), is an important agricultural pest in the Americas and an emerging pest in sub-Saharan Africa, India, East-Asia and Australia, causing damage to major crops such as corn, sorghum and soybean. While... | | Adaptation, Evolutionary Ecology, Expression Studies, Life History, Speciation | Astrid Groot | 2018-05-09 13:04:34 | ||
13 Sep 2019

Deceptive combined effects of short allele dominance and stuttering: an example with Ixodes scapularis, the main vector of Lyme disease in the U.S.A.New curation method for microsatellite markers improves population genetics analysesRecommended by Aurelien Tellier based on reviews by Eric Petit, Martin Husemann and 2 anonymous reviewers based on reviews by Eric Petit, Martin Husemann and 2 anonymous reviewers
Genetic markers are used for in modern population genetics/genomics to uncover the past neutral and selective history of population and species. Besides Single Nucleotide Polymorphisms (SNPs) obtained from whole genome data, microsatellites (or Short Tandem Repeats, SSR) have been common markers of choice in numerous population genetics studies of non-model species with large sample sizes [1]. Microsatellites can be used to uncover and draw inference of the past population demography (e.g. expansion, decline, bottlenecks…), population split, population structure and gene flow, but also life history traits and modes of reproduction (e.g. [2,3]). These markers are widely used in conservation genetics [4] or to study parasites or disease vectors [5]. Microsatellites do show higher mutation rate than SNPs increasing, on the one hand, the statistical power to infer recent events (for example crop domestication, [2,3]), while, on the other hand, decreasing their statistical power over longer time scales due to homoplasy [6]. References [1] Jarne, P., and Lagoda, P. J. (1996). Microsatellites, from molecules to populations and back. Trends in ecology & evolution, 11(10), 424-429. doi: 10.1016/0169-5347(96)10049-5 | Deceptive combined effects of short allele dominance and stuttering: an example with Ixodes scapularis, the main vector of Lyme disease in the U.S.A. | Thierry De Meeûs, Cynthia T. Chan, John M. Ludwig, Jean I. Tsao, Jaymin Patel, Jigar Bhagatwala, and Lorenza Beati | <p>Null alleles, short allele dominance (SAD), and stuttering increase the perceived relative inbreeding of individuals and subpopulations as measured by Wright’s FIS and FST. Ascertainment bias, due to such amplifying problems are usually caused ... | | Evolutionary Ecology, Other, Population Genetics / Genomics | Aurelien Tellier | 2019-05-02 20:52:08 | ||
29 Nov 2022

Joint inference of adaptive and demographic history from temporal population genomic dataInference of genome-wide processes using temporal population genomic dataRecommended by Aurelien Tellier based on reviews by Lawrence Uricchio and 2 anonymous reviewers
Evolutionary genomics, and population genetics in particular, aim to decipher the respective influence of neutral and selective forces shaping genetic polymorphism in a species/population. This is a much-needed requirement before scanning genome data for footprints of species adaptation to their biotic and abiotic environment (Johri et al. 2022). In general, we would like to quantify the proportion of the genome evolving neutrally and under selective (positive, balancing and negative) pressures (Kern and Hahn 2018, Johri et al. 2021). We thus need to understand patterns of linked selection along the genome, that is how the distribution of genetic polymorphisms is shaped by selected sites and the recombination landscape. The present contribution by Pavinato et al. (2022) provides an additional method in the population genomics toolbox to quantify the extent of linked positive and negative selection using temporal data. The availability of genomics data for model and non-model species has led to improvement of the modeling framework for demography and selection (Johri et al. 2022), but also new inference methods making use of the full genome data based on the Sequential Markovian Coalescent (SMC, Li and Durbin 2011), Approximate Bayesian Computation (ABC, Jay et al. 2019), ABC and machine learning (Pudlo et al. 2016, Raynal et al. 2019) or Deep Learning (Sanchez et al. 2021). These methods are based on one sample in time and the use of the coalescent theory to reconstruct the past (demographic) history. However, it is also possible to obtain for many species temporal data sampled over several time points. For species with short generation time (in experimental evolution or monitored populations), one can sample a population every couple of generations as exemplified with Drosophila melanogaster (Bergland et al. 2010). For species with longer generation times that cannot be easily regularly sampled in time, it becomes possible to sequence available specimens from museums (e.g. Cridland et al. 2018) or ancient DNA samples. Methods using temporal data are based on the classical population genomics assumption that demography (migration, population subdivision, population size changes) leaves a genome-wide signal, while selection leaves a localized signal in the close vicinity of the causal mutation. Several methods do assess the demography of a population (change in effective population size, Ne, in time) using temporal data (e.g. Jorde and Ryman 2007) which can be used to calibrate the detection of loci under strong positive selection (Foll et al. 2014). Recently Buffalo and Coop (2020) used genome-wide covariance between allele frequency changes across time samples (and across replicates) to quantify the effects of linked selection over short timescales. In the present contribution, Pavinato et al. (2022) make use of temporal data to draw the joint estimation of demographic and selective parameters using a simulation-based method (ABC-Random Forests). This study by Pavinato et al. (2022) builds a framework allowing to infer the census size of the population in time (N) separately from the effect of genetic drift, which is determined by change in effective population size (Ne) in time, as well estimates of genome-wide parameters of selection. In a nutshell, the authors use a forward simulator and summarize genome data by genomic windows using classic statistics (nucleotide diversity, Tajima’s D, FST, heterozygosity) between time samples and for each sample. They specifically use the distributions (higher moments) of these statistics among all windows. The authors combine as input for the ABC-RF, vectors of summary statistics, model parameters and five latent variables: Ne, the ratio Ne/N, the number of beneficial mutations under strong selection, the average selection coefficient of strongly selected mutations, and the average substitution load. Indeed, the authors are interested in three different types of selection components: 1) the adaptive potential of a population which is estimated as the population mutation rate of beneficial mutations (θb), 2) the number of mutations under strong selection (irrespective of whether they reached fixation or not), and 3) the overall population fitness which is a function of the genetic load. In other words, the novelty of this method is not to focus on the detection of loci under selection, but to infer key parameters/distributions summarizing the genome-wide signal of demography and (positive and negative) selection. As a proof of principle, the authors then apply their method to a dataset of feral populations of honey bees (Apis mellifera) collected in California across many years and recovered from Museum samples (Cridland et al. 2018). The approach yields estimates of Ne which are on the same order of magnitude of previous estimates in hymenopterans, and the authors discuss why the different populations show various values of Ne and N which can be explained by different history of admixture with wild but also domesticated lineages of bees. This study focuses on quantifying the genome-wide joint footprints of demography, and strong positive and negative selection to determine which proportion of the genome evolves neutrally or not. Further application of this method can be anticipated, for example, to study species with ecological and life-history traits which generate discrepancies between census size and Ne, for example for plants with selfing or seed banking (Sellinger et al. 2020), and for which the genome-wide effect of linked selection is not fully understood. References Johri P, Aquadro CF, Beaumont M, Charlesworth B, Excoffier L, Eyre-Walker A, Keightley PD, Lynch M, McVean G, Payseur BA, Pfeifer SP, Stephan W, Jensen JD (2022) Recommendations for improving statistical inference in population genomics. PLOS Biology, 20, e3001669. https://doi.org/10.1371/journal.pbio.3001669 Kern AD, Hahn MW (2018) The Neutral Theory in Light of Natural Selection. Molecular Biology and Evolution, 35, 1366–1371. https://doi.org/10.1093/molbev/msy092 Johri P, Riall K, Becher H, Excoffier L, Charlesworth B, Jensen JD (2021) The Impact of Purifying and Background Selection on the Inference of Population History: Problems and Prospects. Molecular Biology and Evolution, 38, 2986–3003. https://doi.org/10.1093/molbev/msab050 Pavinato VAC, Mita SD, Marin J-M, Navascués M de (2022) Joint inference of adaptive and demographic history from temporal population genomic data. bioRxiv, 2021.03.12.435133, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.03.12.435133 Li H, Durbin R (2011) Inference of human population history from individual whole-genome sequences. Nature, 475, 493–496. https://doi.org/10.1038/nature10231 Jay F, Boitard S, Austerlitz F (2019) An ABC Method for Whole-Genome Sequence Data: Inferring Paleolithic and Neolithic Human Expansions. Molecular Biology and Evolution, 36, 1565–1579. https://doi.org/10.1093/molbev/msz038 Pudlo P, Marin J-M, Estoup A, Cornuet J-M, Gautier M, Robert CP (2016) Reliable ABC model choice via random forests. Bioinformatics, 32, 859–866. https://doi.org/10.1093/bioinformatics/btv684 Raynal L, Marin J-M, Pudlo P, Ribatet M, Robert CP, Estoup A (2019) ABC random forests for Bayesian parameter inference. Bioinformatics, 35, 1720–1728. https://doi.org/10.1093/bioinformatics/bty867 Sanchez T, Cury J, Charpiat G, Jay F (2021) Deep learning for population size history inference: Design, comparison and combination with approximate Bayesian computation. Molecular Ecology Resources, 21, 2645–2660. https://doi.org/10.1111/1755-0998.13224 Bergland AO, Behrman EL, O’Brien KR, Schmidt PS, Petrov DA (2014) Genomic Evidence of Rapid and Stable Adaptive Oscillations over Seasonal Time Scales in Drosophila. PLOS Genetics, 10, e1004775. https://doi.org/10.1371/journal.pgen.1004775 Cridland JM, Ramirez SR, Dean CA, Sciligo A, Tsutsui ND (2018) Genome Sequencing of Museum Specimens Reveals Rapid Changes in the Genetic Composition of Honey Bees in California. Genome Biology and Evolution, 10, 458–472. https://doi.org/10.1093/gbe/evy007 Jorde PE, Ryman N (2007) Unbiased Estimator for Genetic Drift and Effective Population Size. Genetics, 177, 927–935. https://doi.org/10.1534/genetics.107.075481 Foll M, Shim H, Jensen JD (2015) WFABC: a Wright–Fisher ABC-based approach for inferring effective population sizes and selection coefficients from time-sampled data. Molecular Ecology Resources, 15, 87–98. https://doi.org/10.1111/1755-0998.12280 Buffalo V, Coop G (2020) Estimating the genome-wide contribution of selection to temporal allele frequency change. Proceedings of the National Academy of Sciences, 117, 20672–20680. https://doi.org/10.1073/pnas.1919039117 Sellinger TPP, Awad DA, Moest M, Tellier A (2020) Inference of past demography, dormancy and self-fertilization rates from whole genome sequence data. PLOS Genetics, 16, e1008698. https://doi.org/10.1371/journal.pgen.1008698 | Joint inference of adaptive and demographic history from temporal population genomic data | Vitor A. C. Pavinato, Stéphane De Mita, Jean-Michel Marin, Miguel de Navascués | <p style="text-align: justify;">Disentangling the effects of selection and drift is a long-standing problem in population genetics. Simulations show that pervasive selection may bias the inference of demography. Ideally, models for the inference o... | | Adaptation, Population Genetics / Genomics | Aurelien Tellier | 2021-10-20 09:41:26 | ||
18 Jan 2023
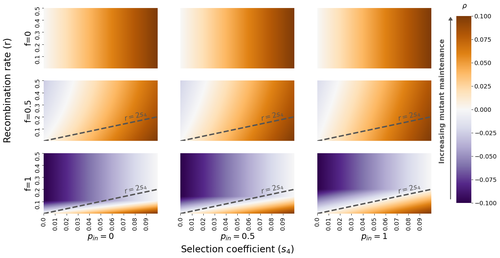
The fate of recessive deleterious or overdominant mutations near mating-type loci under partial selfingMaintenance of deleterious mutations and recombination suppression near mating-type loci under selfingRecommended by Aurelien Tellier based on reviews by 3 anonymous reviewers
The causes and consequences of the evolution of sexual reproduction are a major topic in evolutionary biology. With advances in sequencing technology, it becomes possible to compare sexual chromosomes across species and infer the neutral and selective processes shaping polymorphism at these chromosomes. Most sex and mating-type chromosomes exhibit an absence of recombination in large genomic regions around the animal, plant or fungal sex-determining genes. This suppression of recombination likely occurred in several time steps generating stepwise increasing genomic regions starting around the sex-determining genes. This mechanism generates so-called evolutionary strata of differentiation between sex chromosomes (Nicolas et al., 2004, Bergero and Charlesworth, 2009, Hartmann et al. 2021). The evolution of extended regions of recombination suppression is also documented on mating-type chromosomes in fungi (Hartmann et al., 2021) and around supergenes (Yan et al., 2020, Jay et al., 2021). The exact reason and evolutionary mechanisms for this phenomenon are still, however, debated. Two hypotheses are proposed: 1) sexual antagonism (Charlesworth et al., 2005), which, nevertheless, does explain the observed occurrence of the evolutionary strata, and 2) the sheltering of deleterious alleles by inversions carrying a lower load than average in the population (Charlesworth and Wall, 1999, Antonovics and Abrams, 2004). In the latter, the mechanism is as follows. A genetic inversion or a suppressor of recombination in cis may exhibit some overdominance behaviour. The inversion exhibiting less recessive deleterious mutations (compared to others at the same locus) may increase in frequency, before at higher frequency occurring at the homozygous state, expressing its genetic load. However, the inversion may never be at the homozygous state if it is genetically linked to a gene in a permanently heterozygous state. The inversion can then be advantageous and may reach fixation at the sex chromosome (Charlesworth and Wall, 1999, Antonovics and Abrams, 2004, Jay et al., 2022). These selective mechanisms promote thus the suppression of recombination around the sex-determining gene, and recessive deleterious mutations are permanently sheltered. This hypothesis is corroborated by the sheltering of deleterious mutations observed around loci under balancing selection (Llaurens et al. 2009, Lenz et al. 2016) and around mating-type genes in fungi and supergenes (Jay et al. 2021, Jay et al., 2022). In this present theoretical study, Tezenas et al. (2022) analyse how linkage to a necessarily heterozygous fungal mating type locus influences the persistence/extinction time of a new mutation at a second selected locus. This mutation can either be deleterious and recessive, or overdominant. There is arbitrary linkage between the two loci, and sexual reproduction occurs either between 1) gametes of different individuals (outcrossing), or 2) by selfing with gametes originating from the same (intra-tetrad) or different (inter-tetrad) tetrads produced by that individual. Note, here, that the mating-type gene does not prevent selfing. The authors study the initial stochastic dynamics of the mutation using a multi-type branching process (and simulations when analytical results cannot be obtained) to compute the extinction time of the deleterious mutation. The main result is that the presence of a mating-type locus always decreases the purging probability and increases the purging time of the mutations under selfing. Ultimately, deleterious mutations can indeed accumulate near the mating-type locus over evolutionary time scales. In a nutshell, high selfing or high intra-tetrad mating do increase the sheltering effect of the mating-type locus. In effect, the outcome of sheltering of deleterious mutations depends on two opposing mechanisms: 1) a higher selfing rate induces a greater production of homozygotes and an increased effect of the purging of deleterious mutations, while 2) a higher intra-tetrad selfing rate (or linkage with the mating-type locus) generates heterozygotes which have a small genetic load (and are favoured). The authors also show that rare events of extremely long maintenance of deleterious mutations can occur. The authors conclude by highlighting the manifold effect of selfing which reduces the effective population size and thus impairs the efficiency of selection and increases the mutational load, while also favouring the purge of deleterious homozygous mutations. Furthermore, this study emphasizes the importance of studying the maintenance and accumulation of deleterious mutations in the vicinity of heterozygous loci (e.g. under balancing selection) in selfing species. References Antonovics J, Abrams JY (2004) Intratetrad Mating and the Evolution of Linkage Relationships. Evolution, 58, 702–709. https://doi.org/10.1111/j.0014-3820.2004.tb00403.x Bergero R, Charlesworth D (2009) The evolution of restricted recombination in sex chromosomes. Trends in Ecology & Evolution, 24, 94–102. https://doi.org/10.1016/j.tree.2008.09.010 Charlesworth D, Morgan MT, Charlesworth B (1990) Inbreeding Depression, Genetic Load, and the Evolution of Outcrossing Rates in a Multilocus System with No Linkage. Evolution, 44, 1469–1489. https://doi.org/10.1111/j.1558-5646.1990.tb03839.x Charlesworth D, Charlesworth B, Marais G (2005) Steps in the evolution of heteromorphic sex chromosomes. Heredity, 95, 118–128. https://doi.org/10.1038/sj.hdy.6800697 Charlesworth B, Wall JD (1999) Inbreeding, heterozygote advantage and the evolution of neo–X and neo–Y sex chromosomes. Proceedings of the Royal Society of London. Series B: Biological Sciences, 266, 51–56. https://doi.org/10.1098/rspb.1999.0603 Hartmann FE, Duhamel M, Carpentier F, Hood ME, Foulongne-Oriol M, Silar P, Malagnac F, Grognet P, Giraud T (2021) Recombination suppression and evolutionary strata around mating-type loci in fungi: documenting patterns and understanding evolutionary and mechanistic causes. New Phytologist, 229, 2470–2491. https://doi.org/10.1111/nph.17039 Jay P, Chouteau M, Whibley A, Bastide H, Parrinello H, Llaurens V, Joron M (2021) Mutation load at a mimicry supergene sheds new light on the evolution of inversion polymorphisms. Nature Genetics, 53, 288–293. https://doi.org/10.1038/s41588-020-00771-1 Jay P, Tezenas E, Véber A, Giraud T (2022) Sheltering of deleterious mutations explains the stepwise extension of recombination suppression on sex chromosomes and other supergenes. PLOS Biology, 20, e3001698. https://doi.org/10.1371/journal.pbio.3001698 Lenz TL, Spirin V, Jordan DM, Sunyaev SR (2016) Excess of Deleterious Mutations around HLA Genes Reveals Evolutionary Cost of Balancing Selection. Molecular Biology and Evolution, 33, 2555–2564. https://doi.org/10.1093/molbev/msw127 Llaurens V, Gonthier L, Billiard S (2009) The Sheltered Genetic Load Linked to the S Locus in Plants: New Insights From Theoretical and Empirical Approaches in Sporophytic Self-Incompatibility. Genetics, 183, 1105–1118. https://doi.org/10.1534/genetics.109.102707 Nicolas M, Marais G, Hykelova V, Janousek B, Laporte V, Vyskot B, Mouchiroud D, Negrutiu I, Charlesworth D, Monéger F (2004) A Gradual Process of Recombination Restriction in the Evolutionary History of the Sex Chromosomes in Dioecious Plants. PLOS Biology, 3, e4. https://doi.org/10.1371/journal.pbio.0030004 Tezenas E, Giraud T, Véber A, Billiard S (2022) The fate of recessive deleterious or overdominant mutations near mating-type loci under partial selfing. bioRxiv, 2022.10.07.511119, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.10.07.511119 Yan Z, Martin SH, Gotzek D, Arsenault SV, Duchen P, Helleu Q, Riba-Grognuz O, Hunt BG, Salamin N, Shoemaker D, Ross KG, Keller L (2020) Evolution of a supergene that regulates a trans-species social polymorphism. Nature Ecology & Evolution, 4, 240–249. https://doi.org/10.1038/s41559-019-1081-1 | The fate of recessive deleterious or overdominant mutations near mating-type loci under partial selfing | Emilie Tezenas, Tatiana Giraud, Amandine Veber, Sylvain Billiard | <p style="text-align: justify;">Large regions of suppressed recombination having extended over time occur in many organisms around genes involved in mating compatibility (sex-determining or mating-type genes). The sheltering of deleterious alleles... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Theory, Genome Evolution, Population Genetics / Genomics, Reproduction and Sex | Aurelien Tellier | 2022-10-10 13:50:30 | ||
02 Feb 2024
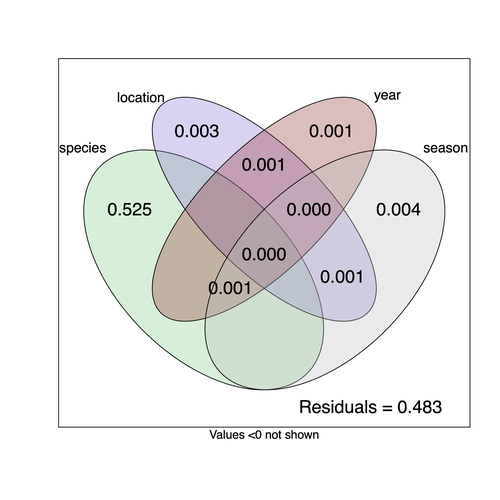
Community structure of heritable viruses in a Drosophila-parasitoids complexThe virome of a Drosophilidae-parasitoid communityRecommended by Ben Longdon based on reviews by 3 anonymous reviewers
Understanding the factors that shape the virome of a host is key to understanding virus ecology and evolution (Obbard, 2018; French & Holmes, 2020). There is still much to learn about the diversity and distribution of viruses in a host community (Wille et al., 2019; Chen et al., 2023). The viruses of parasitoid wasps are well studied, and their viruses, or integrated viral genes, are known to suppress their insect host’s immune response to enhance parasitoid survival (Herniou et al., 2013; Coffman et al., 2022). Likewise, the insect virome is being increasingly well studied (Shi et al., 2016), with the virome of Drosophila species being particularly well characterised over the best part of the last century (L'Heritier & Teissier, 1937; L'Heritier, 1970; Brun & Plus, 1980; Longdon et al., 2010; Longdon et al., 2011; Longdon et al., 2012; Webster et al., 2015; Webster et al., 2016; Medd et al., 2018; Wallace et al., 2021). However, the viromes of parasitoids and their insect host communities have been less well studied (Leigh et al., 2018; Caldas-Garcia et al., 2023), and the inherent connectivity between parasitoids and their hosts provides an interesting system to study virus host range and cross-species transmission. Here, Varaldi et al (Varaldi et al., 2024) have examined the viruses associated with a community of nine Drosophilidae hosts and six parasitoids. Using both RNA and DNA sequencing of insects reared for two generations, they selected viruses that are maintained in the lab either via vertical transmission or contamination of rearing medium. From 55 pools of insects they found 53 virus-like sequences, 37 of which were novel. Parasitoids were host to nearly twice as many viruses as their Drosophila hosts, although they note this could be due to differences in the rearing temperatures of the hosts. They next quantified if species, year, season, or location played a role in structuring the virome, finding only a significant effect of host species, which explained just over 50% of the variation in virus distribution. No evidence was found of related species sharing more similar virus communities. Although looking at a limited number of species, this suggests that these viruses are not co-speciating or preferentially host switching between closely related species. Finally, they carried out crosses between lines of the parasitoid Leptopilina heterotoma that were infected and uninfected for a novel Iflavirus found in their sequencing data. They found evidence of high levels of maternal transmission and lower level horizontal transmission between wasp larvae parasitising the same host. No evidence of changes in parasitoid-induced mortality, developmental success or the sex ratio was found in iflavirus-infected parasitoids. Interestingly individuals infected with this RNA virus also contained viral DNA, but this did not appear to be integrated into the wasp genome. Overall, this work has taken the first steps in examining the community structure of the virome of parasitoids together with their Drosophilidae hosts. This work will not doubt stimulate follow-up studies to explore the evolution and ecology of these novel virus communities. References Brun G, Plus N (1980) The viruses of Drosophila. In: The genetics and biology of Drosophila eds Ashburner M & Wright TRF), pp. 625-702. Academic Press, New York. | Community structure of heritable viruses in a *Drosophila*-parasitoids complex | Julien Varaldi, David Lepetit, Nelly Burlet, Camille Faber, Bérénice Baretje, Roland Allemand | <p style="text-align: justify;">The diversity and phenotypic impacts related to the presence of heritable bacteria in insects have been extensively studied in the last decades. On the contrary, heritable viruses have been overlooked for several re... | | Evolutionary Ecology, Species interactions | Ben Longdon | 2023-08-03 01:07:43 |




