
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields | Recommender▼ | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
18 Dec 2017
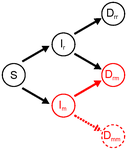
Co-evolution of virulence and immunosuppression in multiple infectionsTwo parasites, virulence and immunosuppression: how does the whole thing evolve?Recommended by Sara Magalhaes based on reviews by 2 anonymous reviewersHow parasite virulence evolves is arguably the most important question in both the applied and fundamental study of host-parasite interactions. Typically, this research area has been progressing through the formalization of the problem via mathematical modelling. This is because the question is a complex one, as virulence is both affected and affects several aspects of the host-parasite interaction. Moreover, the evolution of virulence is a problem in which ecology (epidemiology) and evolution (changes in trait values through time) are tightly intertwined, generating what is now known as eco-evolutionary dynamics. Therefore, intuition is not sufficient to address how virulence may evolve. References [1] Anderson RM and May RM. 1982. Coevolution of hosts and parasites. Parasitology, 1982. 85: 411–426. doi: 10.1017/S0031182000055360 [2] Kamiya T, Mideo N and Alizon S. 2017. Coevolution of virulence and immunosuppression in multiple infections. bioRxiv, ver. 7 peer-reviewed by PCI Evol Biol, 149211. doi: 10.1101/139147 | Co-evolution of virulence and immunosuppression in multiple infections | Tsukushi Kamiya, Nicole Mideo, Samuel Alizon | Many components of the host-parasite interaction have been shown to affect the way virulence, that is parasite induced harm to the host, evolves. However, co-evolution of multiple traits is often neglected. We explore how an immunosuppressive mech... | | Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory | Sara Magalhaes | 2017-06-13 16:49:45 | ||
06 Jun 2019
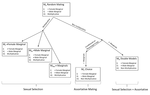
Multi-model inference of non-random mating from an information theoretic approachTell me who you mate with, I’ll tell you what’s going onRecommended by Sara Magalhaes and Alexandre Courtiol based on reviews by Alexandre Courtiol and 2 anonymous reviewersThe study of sexual selection goes as far as Darwin himself. Since then, elaborate theories concerning both intra- and inter-sexual sexual have been developed, and elegant experiments have been designed to test this body of theory. It may thus come as a surprise that the community is still debating on the correct way to measure simple components of sexual selection, such as the Bateman gradient (i.e., the covariance between the number of matings and the number of offspring)[1,2], or to quantify complex behaviours such as mate choice (the non-random choice of individuals with particular characters as mates)[3,4] and their consequences. References [1] Bateman, A. J. (1948). Intra-sexual selection in Drosophila. Heredity, 2(3), 349-368. doi: 10.1038/hdy.1948.21 | Multi-model inference of non-random mating from an information theoretic approach | Antonio Carvajal-Rodríguez | <p>Non-random mating has a significant impact on the evolution of organisms. Here, I developed a modelling framework for discrete traits (with any number of phenotypes) to explore different models connecting the non-random mating causes (mate comp... | | Evolutionary Ecology, Evolutionary Theory, Sexual Selection | Sara Magalhaes | 2019-02-08 19:24:03 | ||
30 Aug 2021
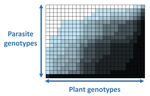
The quasi-universality of nestedness in the structure of quantitative plant-parasite interactionsNestedness and modularity in plant-parasite infection networksRecommended by Santiago Elena based on reviews by Rubén González and 2 anonymous reviewers based on reviews by Rubén González and 2 anonymous reviewers
In a landmark paper, Flores et al. (2011) showed that the interactions between bacteria and their viruses could be nicely described using a bipartite infection networks. Two quantitative properties of these networks were of particular interest, namely modularity and nestedness. Modularity emerges when groups of host species (or genotypes) shared groups of viruses. Nestedness provided a view of the degree of specialization of both partners: high nestedness suggests that hosts differ in their susceptibility to infection, with some highly susceptible host genotypes selecting for very specialized viruses while strongly resistant host genotypes select for generalist viruses. Translated to the plant pathology parlance, this extreme case would be equivalent to a gene-for-gene infection model (Flor 1956): new mutations confer hosts with resistance to recently evolved viruses while maintaining resistance to past viruses. Likewise, virus mutations for expanding host range evolve without losing the ability to infect ancestral host genotypes. By contrast, a non-nested network would represent a matching-allele infection model (Frank 2000) in which each interacting organism evolves by losing its capacity to resist/infect its ancestral partners, resembling a Red Queen dynamic. Obviously, the reality is more complex and may lie anywhere between these two extreme situations. Recently, Valverde et al. (2020) developed a model to explain the emergence of nestedness and modularity in plant-virus infection networks across diverse habitats. They found that local modularity could coexist with global nestedness and that intraspecific competition was the main driver of the evolution of ecosystems in a continuum between nested-modular and nested networks. These predictions were tested with field data showing the association between plant host species and different viruses in different agroecosystems (Valverde et al. 2020). The effect of interspecific competition in the structure of empirical plant host-virus infection networks was also tested by McLeish et al. (2019). Besides data from agroecosystems, evolution experiments have also shown the pervasive emergence of nestedness during the diversification of independently-evolved lineages of potyviruses in Arabidopsis thaliana genotypes that differ in their susceptibility to infection (Hillung et al. 2014; González et al. 2019; Navarro et al. 2020). In their study, Moury et al. (2021) have expanded all these previous observations to a diverse set of pathosystems that range from viruses, bacteria, oomycetes, fungi, nematodes to insects. While modularity was barely seen in only a few of the systems, nestedness was a common trend (observed in ~94% of all systems). This nestedness, as seen in previous studies and as predicted by theory, emerged as a consequence of the existence of generalist and specialist strains of the parasites that differed in their capacity to infect more or less resistant plant genotypes. As pointed out by Moury et al. (2021) in their conclusions, the ubiquity of nestedness in plant-parasite infection matrices has strong implications for the evolution and management of infectious diseases. References Flor, H. H. (1956). The complementary genic systems in flax and flax rust. In Advances in genetics, 8, 29-54. https://doi.org/10.1016/S0065-2660(08)60498-8 Flores, C. O., Meyer, J. R., Valverde, S., Farr, L., and Weitz, J. S. (2011). Statistical structure of host–phage interactions. Proceedings of the National Academy of Sciences, 108, E288-E297. https://doi.org/10.1073/pnas.1101595108 Frank, S. A. (2000). Specific and non-specific defense against parasitic attack. Journal of Theoretical Biology, 202, 283-304. https://doi.org/10.1006/jtbi.1999.1054 González, R., Butković, A., and Elena, S. F. (2019). Role of host genetic diversity for susceptibility-to-infection in the evolution of virulence of a plant virus. Virus evolution, 5(2), vez024. https://doi.org/10.1093/ve/vez052 Hillung, J., Cuevas, J. M., Valverde, S., and Elena, S. F. (2014). Experimental evolution of an emerging plant virus in host genotypes that differ in their susceptibility to infection. Evolution, 68, 2467-2480. https://doi.org/10.1111/evo.12458 McLeish, M., Sacristán, S., Fraile, A., and García-Arenal, F. (2019). Coinfection organizes epidemiological networks of viruses and hosts and reveals hubs of transmission. Phytopathology, 109, 1003-1010. https://doi.org/10.1094/PHYTO-08-18-0293-R Moury B, Audergon J-M, Baudracco-Arnas S, Krima SB, Bertrand F, Boissot N, Buisson M, Caffier V, Cantet M, Chanéac S, Constant C, Delmotte F, Dogimont C, Doumayrou J, Fabre F, Fournet S, Grimault V, Jaunet T, Justafré I, Lefebvre V, Losdat D, Marcel TC, Montarry J, Morris CE, Omrani M, Paineau M, Perrot S, Pilet-Nayel M-L and Ruellan Y (2021) The quasi-universality of nestedness in the structure of quantitative plant-parasite interactions. bioRxiv, 2021.03.03.433745, ver. 4 recommended and peer-reviewed by PCI Evolutionary Biology. https://doi.org/10.1101/2021.03.03.433745 Navarro, R., Ambros, S., Martinez, F., Wu, B., Carrasco, J. L., and Elena, S. F. (2020). Defects in plant immunity modulate the rates and patterns of RNA virus evolution. bioRxiv. doi: https://doi.org/10.1101/2020.10.13.337402 Valverde, S., Vidiella, B., Montañez, R., Fraile, A., Sacristán, S., and García-Arenal, F. (2020). Coexistence of nestedness and modularity in host–pathogen infection networks. Nature ecology & evolution, 4, 568-577. https://doi.org/10.1038/s41559-020-1130-9 | The quasi-universality of nestedness in the structure of quantitative plant-parasite interactions | Moury Benoît, Audergon Jean-Marc, Baudracco-Arnas Sylvie, Ben Krima Safa, Bertrand François, Boissot Nathalie, Buisson Mireille, Caffier Valérie, Cantet Mélissa, Chanéac Sylvia, Constant Carole, Delmotte François, Dogimont Catherine, Doumayrou Jul... | <p>Understanding the relationships between host range and pathogenicity for parasites, and between the efficiency and scope of immunity for hosts are essential to implement efficient disease control strategies. In the case of plant parasites, most... | | Bioinformatics & Computational Biology, Evolutionary Dynamics, Species interactions | Santiago Elena | 2021-03-04 21:23:08 | ||
04 Mar 2024
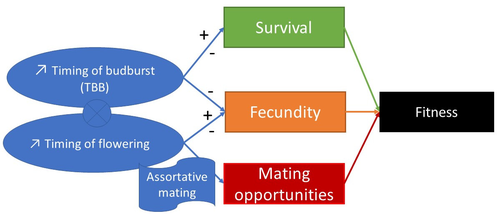
Interplay between fecundity, sexual and growth selection on the spring phenology of European beech (Fagus sylvatica L.).Interplay between fecundity, sexual and growth selection on the spring phenology of European beech (Fagus sylvatica L.)Recommended by Santiago C. Gonzalez-Martinez based on reviews by 2 anonymous reviewers
Starting with the seminar paper by Lande & Arnold (1983), several studies have addressed phenotypic selection in natural populations of a wide variety of organisms, with a recent renewed interest in forest trees (e.g., Oddou-Muratorio et al. 2018; Alexandre et al. 2020; Westergren et al. 2023). Because of their long generation times, long-lived organisms such as forest trees may suffer the most from maladaptation due to climate change, and whether they will be able to adapt to new environmental conditions in just one or a few generations is hotly debated. In this study, Oddou-Muratorio and colleagues (2024) extend the current framework to add two additional selection components that may alter patterns of fecundity selection and the estimation of standard selection gradients, namely sexual selection (evaluated as differences in flowering phenology conducting to assortative mating) and growth (viability) selection. Notably, the study is conducted in two contrasted environments (low vs high altitude populations) providing information on how the environment may modulate selection patterns in spring phenology. Spring phenology is a key adaptive trait that has been shown to be already affected by climate change in forest trees (Alberto et al. 2013). While fecundity selection for early phenology has been extensively reported before (see Munguía-Rosas et al. 2011), the authors found that this kind of selection can be strongly modulated by sexual selection, depending on the environment. Moreover, they found a significant correlation between early phenology and seedling growth in a common garden, highlighting the importance of this trait for early survival in European beech. As a conclusion, this original research puts in evidence the need for more integrative approaches for the study of natural selection in the field, as well as the importance of testing multiple environments and the relevance of common gardens to further evaluate phenotypic changes due to real-time selection. PS: The recommender and the first author of the preprint have shared authorship in a recent paper in a similar topic (Westergren et al. 2023). Nevertheless, the recommender has not contributed in any way or was aware of the content of the current preprint before acting as recommender, and steps have been taken for a fair and unpartial evaluation. References Alberto, F. J., Aitken, S. N., Alía, R., González‐Martínez, S. C., Hänninen, H., Kremer, A., Lefèvre, F., Lenormand, T., Yeaman, S., Whetten, R., & Savolainen, O. (2013). Potential for evolutionary responses to climate change - evidence from tree populations. Global Change Biology, 19(6), 1645‑1661. Oddou-Muratorio S, Bontemps A, Gauzere J, Klein E (2024) Interplay between fecundity, sexual and growth selection on the spring phenology of European beech (Fagus sylvatica L.). bioRxiv, 2023.04.27.538521, ver. 2 peer-reviewed and recommended by Peer Community In Evolutionary Biology https://doi.org/10.1101/2023.04.27.538521 Oddou-Muratorio, S., Gauzere, J., Bontemps, A., Rey, J.-F., & Klein, E. K. (2018). Tree, sex and size: Ecological determinants of male vs. female fecundity in three Fagus sylvatica stands. Molecular Ecology, 27(15), 3131‑3145. | Interplay between fecundity, sexual and growth selection on the spring phenology of European beech (*Fagus sylvatica* L.). | Sylvie Oddou-Muratorio, Aurore Bontemps, Julie Gauzere, Etienne Klein | <p>Background: Plant phenological traits such as the timing of budburst or flowering can evolve on ecological timescales through response to fecundity and viability selection. However, interference with sexual selection may arise from assortative ... | | Adaptation, Evolutionary Ecology, Quantitative Genetics, Reproduction and Sex, Sexual Selection | Santiago C. Gonzalez-Martinez | 2023-05-02 11:57:23 | ||
17 Dec 2016
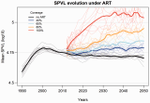
POSTPRINT
Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling studyPredicting HIV virulence evolution in response to widespread treatmentRecommended by Samuel Alizon and Roger Kouyos
It is a classical result in the virulence evolution literature that treatments decreasing parasite replication within the host should select for higher replication rates, thus driving increased levels of virulence if the two are correlated. There is some evidence for this in vitro but very little in the field. HIV infections in humans offer a unique opportunity to go beyond the simple predictions that treatments should favour more virulent strains because many details of this host-parasite system are known, especially the link between set-point virus load, transmission rate and virulence. To tackle this question, Herbeck et al. [1] used a detailed individual-based model. This is original because it allows them to integrate existing knowledge from the epidemiology and evolution of HIV (e.g. recent estimates of the ‘heritability’ of set-point virus load from one infection to the next). This detailed model allows them to formulate predictions regarding the effect of different treatment policies; especially regarding the current policy switch away from treatment initiation based on CD4 counts towards universal treatment. The results show that, perhaps as expected from the theory, treatments based on the level of remaining host target cells (CD4 T cells) do not affect virulence evolution because they do not strongly affect the virulence level that maximizes HIV’s transmission potential. However, early treatments can lead to moderate increase in virulence within several years if coverage is high enough. These results seem quite robust to variation of all the parameters in realistic ranges. The great step forward in this model is the ability to obtain quantitative prediction regarding how a virus may evolve in response to public health policies. Here the main conclusion is that given our current knowledge in HIV biology, the risk of virulence evolution is perhaps more limited than expected from a direct application of virulence evolution model. Interestingly, the authors also conclude that recently observed increased in HIV virulence [2-3] cannot be explained by the impact of antiretroviral therapy alone; which raises the question about the main mechanism behind this increase. Finally, the authors make the interesting suggestion that “changing virulence is amenable to being monitored alongside transmitted drug resistance in sentinel surveillance”. References [1] Herbeck JT, Mittler JE, Gottlieb GS, Goodreau SM, Murphy JT, Cori A, Pickles M, Fraser C. 2016. Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling study. Virus Evolution 2:vew028. doi: 10.1093/ve/vew028 [2] Herbeck JT, Müller V, Maust BS, Ledergerber B, Torti C, et al. 2012. Is the virulence of HIV changing? A meta-analysis of trends in prognostic markers of HIV disease progression and transmission. AIDS 26:193-205. doi: 10.1097/QAD.0b013e32834db418 [3] Pantazis N, Porter K, Costagliola D, De Luca A, Ghosn J, et al. 2014. Temporal trends in prognostic markers of HIV-1 virulence and transmissibility: an observational cohort study. Lancet HIV 1:e119-26. doi: 10.1016/s2352-3018(14)00002-2 | Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling study | Herbeck JT, Mittler JE, Gottlieb GS, Goodreau SM, Murphy JT, Cori A, Pickles M, Fraser C | There are global increases in the use of HIV antiretroviral therapy (ART), guided by clinical benefits of early ART initiation and the efficacy of treatment as prevention of transmission. Separately, it has been shown theoretically and empirically... | | Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Epidemiology | Samuel Alizon | 2016-12-16 20:54:08 | ||
12 Jun 2017

Evolution and manipulation of vector host choiceModelling the evolution of how vector-borne parasites manipulate the vector's host choiceRecommended by Samuel Alizon based on reviews by Samuel Alizon and Nicole Mideo
Many parasites can manipulate their hosts, thus increasing their transmission to new hosts [1]. This is particularly the case for vector-borne parasites, which can alter the feeding behaviour of their hosts. However, predicting the optimal strategy is not straightforward because three actors are involved and the interests of the parasite may conflict with that of the vector. There are few models that consider the evolution of host manipulation by parasites [but see 2-4], but there are virtually none that investigated how parasites can manipulate the host choice of vectors. Even on the empirical side, many aspects of this choice remain unknown. Gandon [5] develops a simple evolutionary epidemiology model that allows him to formulate clear and testable predictions. These depend on which actor controls the trait (the vector or the parasite) and, when there is manipulation, whether it is realised via infected hosts (to attract vectors) or infected vectors (to change host choice). In addition to clarifying the big picture, Gandon [5] identifies some nice properties of the model, for instance an independence of the density/frequency-dependent transmission assumption or a backward bifurcation at R0=1, which suggests that parasites could persist even if their R0 is driven below unity. Overall, this study calls for further investigation of the different scenarios with more detailed models and experimental validation of general predictions. References [1] Hughes D, Brodeur J, Thomas F. 2012. Host manipulation by parasites. Oxford University Press. [2] Brown SP. 1999. Cooperation and conflict in host-manipulating parasites. Proceedings of the Royal Society of London B: Biological Sciences 266: 1899–1904. doi: 10.1098/rspb.1999.0864 [3] Lion S, van Baalen M, Wilson WG. 2006. The evolution of parasite manipulation of host dispersal. Proceedings of the Royal Society of London B: Biological Sciences. 273: 1063–1071. doi: 10.1098/rspb.2005.3412 [4] Vickery WL, Poulin R. 2010. The evolution of host manipulation by parasites: a game theory analysis. Evolutionary Ecology 24: 773–788. doi: 10.1007/s10682-009-9334-0 [5] Gandon S. 2017. Evolution and manipulation of vector host choice. bioRxiv 110577, ver. 3 of 7th June 2017. doi: 10.1101/110577 | Evolution and manipulation of vector host choice | Sylvain Gandon | The transmission of many animal and plant diseases relies on the behavior of arthropod vectors. In particular, the choice to feed on either infected or uninfected hosts can dramatically affect the epidemiology of vector-borne diseases. I develop a... | | Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory | Samuel Alizon | 2017-03-03 19:18:54 | ||
09 Feb 2018
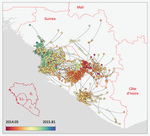
Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreakSimulating the effect of public health interventions using dated virus sequences and geographical dataRecommended by Samuel Alizon based on reviews by Christian Althaus, Chris Wymant and 1 anonymous reviewer
Perhaps because of its deadliness, the 2013-2016 Ebola Virus (EBOV) epidemics in West-Africa has led to unprecedented publication and sharing of full virus genome sequences. This was both rapid (90 full genomes were shared within weeks [1]) and important (more than 1500 full genomes have been released overall [2]). Furthermore, the availability of the metadata (especially GPS location) has led to depth analyses of the geographical spread of the epidemics [3]. References [1] Gire et al. 2014. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345: 1369–1372. doi: 10.1126/science.1259657. | Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreak | Simon Dellicour, Guy Baele, Gytis Dudas, Nuno R. Faria, Oliver G. Pybus, Marc A. Suchard, Andrew Rambaut, Philippe Lemey | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (https://doi.org/10.24072/pci.evolbiol.100046). The recent Ebola virus (EBOV) outbreak in West Africa witnessed considerable efforts to obtain viral genom... | | Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Samuel Alizon | 2017-09-30 13:49:57 | ||
19 Jul 2021

Host phenology can drive the evolution of intermediate virulence strategies in some obligate-killer parasitesModelling parasitoid virulence evolution with seasonalityRecommended by Samuel Alizon based on reviews by Alex Best and 2 anonymous reviewers
The harm most parasites cause to their host, i.e. the virulence, is a mystery because host death often means the end of the infectious period. For obligate killer parasites, or “parasitoids”, that need to kill their host to transmit to other hosts the question is reversed. Indeed, more rapid host death means shorter generation intervals between two infections and mathematical models show that, in the simplest settings, natural selection should always favour more virulent strains (Levin and Lenski, 1983). Adding biological details to the model modifies this conclusion and, for instance, if the relationship between the infection duration and the number of parasites transmission stages produced in a host is non-linear, strains with intermediate levels of virulence can be favoured (Ebert and Weisser 1997). Other effects, such as spatial structure, could yield similar effects (Lion and van Baalen, 2007). In their study, MacDonald et al. (2021) explore another type of constraint, which is seasonality. Earlier studies, such as that by Donnelly et al. (2013) showed that this constraint can affect virulence evolution but they had focused on directly transmitted parasites. Using a mathematical model capturing the dynamics of a parasitoid, MacDonald et al. (2021) show if two main assumptions are met, namely that at the end of the season only transmission stages (or “propagules”) survive and that there is a constant decay of these propagules with time, then strains with intermediate levels of virulence are favoured. Practically, the authors use delay differential equations and an adaptive dynamics approach to identify evolutionary stable strategies. As expected, the longer the short the season length, the higher the virulence (because propagule decay matters less). The authors also identify a non-linear relationship between the variation in host development time and virulence. Generally, the larger the variation, the higher the virulence because the parasitoid has to kill its host before the end of the season. However, if the variation is too wide, some hosts become physically impossible to use for the parasite, whence a decrease in virulence. Finally, MacDonald et ali. (2021) show that the consequence of adding trade-offs between infection duration and the number of propagules produced is in line with earlier studies (Ebert and Weisser 1997). These mathematical modelling results provide testable predictions for using well-described systems in evolutionary ecology such as daphnia parasitoids, baculoviruses, or lytic phages. Reference Donnelly R, Best A, White A, Boots M (2013) Seasonality selects for more acutely virulent parasites when virulence is density dependent. Proc R Soc B, 280, 20122464. https://doi.org/10.1098/rspb.2012.2464 Ebert D, Weisser WW (1997) Optimal killing for obligate killers: the evolution of life histories and virulence of semelparous parasites. Proc R Soc B, 264, 985–991. https://doi.org/10.1098/rspb.1997.0136 Levin BR, Lenski RE (1983) Coevolution in bacteria and their viruses and plasmids. In: Futuyma DJ, Slatkin M eds. Coevolution. Sunderland, MA, USA: Sinauer Associates, Inc., 99–127. Lion S, van Baalen M (2008) Self-structuring in spatial evolutionary ecology. Ecol. Lett., 11, 277–295. https://doi.org/10.1111/j.1461-0248.2007.01132.x MacDonald H, Akçay E, Brisson D (2021) Host phenology can drive the evolution of intermediate virulence strategies in some obligate-killer parasites. bioRxiv, 2021.03.13.435259, ver. 8 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.03.13.435259 | Host phenology can drive the evolution of intermediate virulence strategies in some obligate-killer parasites | Hannelore MacDonald, Erol Akçay, Dustin Brisson | <p style="text-align: justify;">The traditional mechanistic trade-offs resulting in a negative correlation between transmission and virulence are the foundation of nearly all current theory on the evolution of parasite virulence. Several ecologica... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory | Samuel Alizon | 2021-03-14 13:47:33 | ||
05 Oct 2017

Using Connectivity To Identify Climatic Drivers Of Local AdaptationA new approach to identifying drivers of local adaptationRecommended by Ruth Arabelle Hufbauer based on reviews by Ruth Arabelle Hufbauer and Thomas LenormandLocal adaptation, the higher fitness a population achieves in its local “home” environment relative to other environments is a crucial phase in the divergence of populations, and as such both generates and maintains diversity. Local adaptation is enhanced by selection and genetic variation in the relevant traits, and decreased by gene flow and genetic drift. Demonstrating local adaptation is laborious, and is typically done with a reciprocal transplant design [1], documenting repeated geographic clines [e.g. 2, 3] also provides strong evidence of local adaptation. Even when well documented, it is often unknown which aspects of the environment impose selection. Indeed, differences in environment between different sites that are measured during studies of local adaptation explain little of the variance in the degree of local adaptation [4]. This poses a problem to population management. Given climate change and habitat destruction, understanding the environmental drivers of local adaptation can be crucially important to conducting successful assisted migration or targeted gene flow. In this manuscript, Macdonald et al. [5] propose a means of identifying which aspects of the environment select for local adaptation without conducting a reciprocal transplant experiment. The idea is that the strength of relationships between traits and environmental variables that are due to plastic responses to the environment will not be influenced by gene flow, but the strength of trait-environment relationships that are due to local adaptation should decrease with gene flow. This then can be used to reduce the somewhat arbitrary list of environmental variables on which data are available down to a targeted list more likely to drive local adaptation in specific traits. To perform such an analysis requires three things: 1) measurements of traits of interest in a species across locations, 2) an estimate of gene flow between locations, which can be replaced with a biologically meaningful estimate of how well connected those locations are from the point of view of the study species, and 3) data on climate and other environmental variables from across a species’ range, many of which are available on line. Macdonald et al. [5] demonstrate their approach using a skink (Lampropholis coggeri). They collected morphological and physiological data on individuals from multiple populations. They estimated connectivity among those locations using information on habitat suitability and dispersal potential [6], and gleaned climatic data from available databases and the literature. They find that two physiological traits, the critical minimum and maximum temperatures, show the strongest signs of local adaptation, specifically local adaptation to annual mean precipitation, precipitation of the driest quarter, and minimum annual temperature. These are then aspects of skink phenotype and skink habitats that could be explored further, or could be used to provide background information if migration efforts, for example for genetic rescue [7] were initiated. The approach laid out has the potential to spark a novel genre of research on local adaptation. It its simplest form, knowing that local adaptation is eroded by gene flow, it is intuitive to consider that if connectivity reduces the strength of the relationship between an environmental variable and a trait, that the trait might be involved in local adaptation. The approach is less intuitive than that, however – it relies not connectivity per-se, but the interaction between connectivity and different environmental variables and how that interaction alters trait-environment relationships. The authors lay out a number of useful caveats and potential areas that could use further development. It will be interesting to see how the community of evolutionary biologists responds. References [1] Blanquart F, Kaltz O, Nuismer SL and Gandon S. 2013. A practical guide to measuring local adaptation. Ecology Letters, 16: 1195-1205. doi: 10.1111/ele.12150 [2] Huey RB, Gilchrist GW, Carlson ML, Berrigan D and Serra L. 2000. Rapid evolution of a geographic cline in size in an introduced fly. Science, 287: 308-309. doi: 10.1126/science.287.5451.308 [3] Milesi P, Lenormand T, Lagneau C, Weill M and Labbé P. 2016. Relating fitness to long-term environmental variations in natura. Molecular Ecology, 25: 5483-5499. doi: 10.1111/mec.13855 [4] Hereford, J. 2009. A quantitative survey of local adaptation and fitness trade-offs. The American Naturalist 173: 579-588. doi: 10.1086/597611 [5] Macdonald SL, Llewelyn J and Phillips BL. 2017. Using connectivity to identify climatic drivers of local adaptation. bioRxiv, ver. 4 of October 4, 2017. doi: 10.1101/145169 [6] Macdonald SL, Llewelyn J, Moritz C and Phillips BL. 2017. Peripheral isolates as sources of adaptive diversity under climate change. Frontiers in Ecology and Evolution, 5:88. doi: 10.3389/fevo.2017.00088 [7] Whiteley AR, Fitzpatrick SW, Funk WC and Tallmon DA. 2015. Genetic rescue to the rescue. Trends in Ecology & Evolution, 30: 42-49. doi: 10.1016/j.tree.2014.10.009 | Using Connectivity To Identify Climatic Drivers Of Local Adaptation | Stewart L. Macdonald, John Llewelyn, Ben Phillips | Despite being able to conclusively demonstrate local adaptation, we are still often unable to objectively determine the climatic drivers of local adaptation. Given the rapid rate of global change, understanding the climatic drivers of local adapta... | | Adaptation, Evolutionary Applications | Ruth Arabelle Hufbauer | Thomas Lenormand | 2017-06-06 13:06:54 | |
31 Jul 2017

Selection on morphological traits and fluctuating asymmetry by a fungal parasite in the yellow dung flyParasite-mediated selection promotes small body size in yellow dung fliesRecommended by Rodrigo Medel based on reviews by Rodrigo Medel and 1 anonymous reviewerBody size has long been considered as one of the most important organismic traits influencing demographical processes, population size, and evolution of life history strategies [1, 2]. While many studies have reported a selective advantage of large body size, the forces that determine small-sized organisms are less known, and reports of negative selection coefficients on body size are almost absent at present. This lack of knowledge is unfortunate as climate change and energy demands in stressful environments, among other factors, may produce new selection scenarios and unexpected selection surfaces [3]. In this manuscript, Blanckenhorn [4] reports on a potential explanation for the surprising 10% body size decrease observed in a Swiss population of yellow dung flies during 1993 - 2009. The author took advantage of a fungus outbreak in 2002 to assess the putative role of the fungus Entomopthora scatophagae, a specific parasite of adult yellow dung flies, as selective force acting upon host body size. His findings indicate that, as expected by sexual selection theory, large males experience a mating advantage. However, this positive sexual selection is opposed by a strong negative selection on male and female body size through the viability fitness component. This study provides the first evidence of parasite-mediated disadvantage of large adult body size in the field. While further experimental work is needed to elucidate the exact causes of body size reduction in the population, the author proposes a variation of the trade-off hypothesis raised by Rantala & Roff [5] that large-sized individuals face an immunity cost due to their high absolute energy demands in stressful environments. References [1] Peters RH. 1983. The ecological implications of body size. Cambridge University Press, Cambridge. [2] Schmidt-Nielsen K. 1984. Scaling: why is animal size so important? Cambridge University Press, Cambridge. [3] Ohlberger J. 2013. Climate warming and ectotherm body size: from individual physiology to community ecology. Functional Ecology 27: 991-1001. doi: 10.1111/1365-2435.12098 [4] Blanckenhorn WU. 2017. Selection on morphological traits and fluctuating asymmetry by a fungal parasite in the yellow dung fly. bioRxiv 136325, ver. 2 of 29th June 2017. doi: 10.1101/136325 [5] Rantala MJ & Roff DA. 2005. An analysis of trade-offs in immune function, body size and development time in the Mediterranean field cricket, Gryllus bimaculatus. Functional Ecology 19: 323-330. doi: 10.1111/j.1365-2435.2005.00979.x | Selection on morphological traits and fluctuating asymmetry by a fungal parasite in the yellow dung fly | Wolf U. Blanckenhorn | Evidence for selective disadvantages of large body size remains scarce in general. Previous phenomenological studies of the yellow dung fly *Scathophaga stercoraria* have demonstrated strong positive sexual and fecundity selection on male and fema... | | Behavior & Social Evolution, Evolutionary Ecology, Life History, Sexual Selection | Rodrigo Medel | Rodrigo Medel | 2017-05-10 11:16:26 |




