
An evolutionary view of a biomedically important gene family
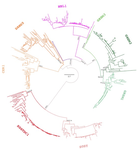
Evolution of the DAN gene family in vertebrates
Abstract
Recommendation: posted 23 July 2020, validated 27 July 2020
Makova, K. (2020) An evolutionary view of a biomedically important gene family. Peer Community in Evolutionary Biology, 100104. https://doi.org/10.24072/pci.evolbiol.100104
Recommendation
This manuscript [1] investigates the evolutionary history of the DAN gene family—a group of genes important for embryonic development of limbs, kidneys, and left-right axis speciation. This gene family has also been implicated in a number of diseases, including cancer and nephropathies. DAN genes have been associated with the inhibition of the bone morphogenetic protein (BMP) signaling pathway. Despite this detailed biochemical and functional knowledge and clear importance for development and disease, evolution of this gene family has remained understudied. The diversification of this gene family was investigated in all major groups of vertebrates. The monophyly of the gene members belonging to this gene family was confirmed. A total of five clades were delineated, and two novel lineages were discovered. The first lineage was only retained in cephalochordates (amphioxus), whereas the second one (GREM3) was retained by cartilaginous fish, holostean fish, and coelanth. Moreover, the patterns of chromosomal synteny in the chromosomal regions harboring DAN genes were investigated. Additionally, the authors reconstructed the ancestral gene repertoires and studied the differential retention/loss of individual gene members across the phylogeny. They concluded that the ancestor of gnathostome vertebrates possessed eight DAN genes that underwent differential retention during the evolutionary history of this group. During radiation of vertebrates, GREM1, GREM2, SOST, SOSTDC1, and NBL1 were retained in all major vertebrate groups. At the same time, GREM3, CER1, and DAND5 were differentially lost in some vertebrate lineages. At least two DAN genes were present in the common ancestor of vertebrates, and at least three DAN genes were present in the common ancestor of chordates. Therefore the patterns of retention and diversification in this gene family appear to be complex. Evolutionary slowdown for the DAN gene family was observed in mammals, suggesting selective constraints. Overall, this article puts the biomedical importance of the DAN family in the evolutionary perspective.
References
[1] Opazo JC, Hoffmann FG, Zavala K, Edwards SV (2020) Evolution of the DAN gene family in vertebrates. bioRxiv, 794404, ver. 3 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/794404

The recommender in charge of the evaluation of the article and the reviewers declared that they have no conflict of interest (as defined in the code of conduct of PCI) with the authors or with the content of the article. The authors declared that they comply with the PCI rule of having no financial conflicts of interest in relation to the content of the article.
no declaration
Evaluation round #1
DOI or URL of the preprint: 10.1101/794404
Version of the preprint: 1
Author's Reply, 11 Jun 2020
Decision by Kateryna Makova, posted 14 Jan 2020
Please revise the manuscript according to the reviewers' suggestions.
Reviewed by anonymous reviewer 2, 13 Jan 2020
Review for "Evolution of the DAN gene family in vertebrates"
In this manuscript by Opazzo et al., the authors use homology searches to identify genes from the DAN gene family (Differential screening-selected gene Aberrant in Neuroblastoma) across chordate lineages. The phylogenetic relationships of these genes were inferred and the toplogy of the resulting tree was used to describe the evolutionary history of the gene family.
Interestingly, the authors identify a new family member related to the Gremlin genes, which they dub Grem3. Next, in the Gnathostome lineage, the authors show evidence for five genes being present in its MRCA that are also widely retained across its descendents (e.g. the major Gnathosome lineages listed in figure 4). These genes include Grem1, Grem2, SOST, SOSTDC1, and NBL1. The authors also identify 3 gene family members that they conclude are likely in the gnathostome ancestor, but have experienced loss in some of the ancestors: Grem3, Cer1, and DAND5.
Over all, the manuscript is well-written and lays out its case fairly well. And for the most part, I find the major arguments to be reasonable. However, there are a lot of areas that I feel would benefit from feedback described here.
Major comments
The methods are insufficiently detailed to permit the work to be repeated
The authors do not define the pool of sequences from which query and subject sequences are drawn. The specific implementation of blast and its version isn't cited. The filtering criteria used to determine whether hits are retained or discarded are not documented. The nature of the multiple alignment wasn't described. How much of the genes were alignable at the greatest divergences? In the introduction, the authors claim that there is "low inter-parallog conservation", indicating that the alignment may not be reliable in many regions. What was aligned? Nucleotides or amino acids (I assume amino acids)?
The results are fairly sparse on details
For example, display items aren't thoroughly described. The captions are very terse. For example, there appears to be a convention in the synteny plots where the absence of a bar indicates the absense of the gene (ag CER1 in Spotted Gar in Figure 2B). However, in Figures 5 and 6, dotted lines apparently indicate missing DAN genes but missing bars for flanking genes means that the gene isn't in the syntenic region. What is the scale in Figure 1? A bar with the number "0.7" is included. The caption doesn't elaborate. I'm accustomed to bootstrap support to be reported in Numerator/Denominator or explicity in %. The numbers corresponding to bootstrap support in Figure 1 are just bare integers.
The authors often point out disagreements with the literature, which is commendable. However, little effort is made to reconcile these observed disagreements. I'd feel better if the authors would discuss the discrepancies they point out.
Examples:
"Although the study of Walsh et al. (2010) supports..., two other studies report alternative topologies."
"Nolan et al. (2014) recovered NBL1 as sister... However, in support of our study Avsian-Kretchmer et al. (2004) recovered NBL1 as sister to the GREM lineages."
"However, in contrast to Petillon et al. (2013), we did not find..."The claim of "recovering monophyly" is confusing to me.
"Our results recovered monophyly of all DAN gene family members"My parsing of this statement in the abstract (and others like it throughout the manuscript) is probably not what the authors intended. To me, this sounds like "we confirmed that, as a group, all DAN genes are monophyletic". This doesn't make sense in an analysis where the recovery of a gene from EnsEMBL is viewed as conferring DAN membership on that gene. So, by definition, every gene in the analysis is DAN, and with no non-DAN genes for contrast, no determination about monophyly can be made.
While I can't confidently interpolate what the authors actually meant, perhaps the following is closer to the authors' meaning:
"For each member of the gene family (e.g. CER1, SOST, SOSTDC1, DAND5, NBL1, GREM1, GREM2, and a new member, GREM5), the group of species sequences corresponding to each gene is monophyletic."
Even this formulation is a bit confusing to me, as the monophyly seems to be how the authors would assign a particular sequence in a particular species to particular family member. And in any event, this gets a bit muddied when there is gene duplication. What is monophyly when for some taxa, there are duplicates, and others, there aren't? Is "recovery of monophyly" a result as implied by the authors? Or rather is it part of how the authors are classifying the sequences into family members like CER1, etc.?
Perhaps this "recovery of monophyly" could be reconciled if the authors inferred the full duplication history with synteny for every species they examined and then layered the phylogenetic analysis of the gene family on top of that. But, as far as I can tell, this was not the strategy the authors followed in most cases.
Finally, DAND5 doesn't appear to offer strong support for monophyly given that lack of support for placing the Coelacanth as sister to the other DAND5 genes. The strong synteny argument doesn't change this assessment, as it could be a brute fact that the Coelacanth sequence is simultaneously the DAND5 ortholog and there is no strong evidence of monophyly with the remaining DAND5 orthologs.
One comment relating to paralogy confused me.
"The fourth clade corresponds to the NBL1 gene, the founding member of the DAN gene family, and was recovered as monophyletic with strong support (pink clade; Fig. 1)."
This way of discussing paralogy (ie "founding member") seems clumsy to me. Barring clear mechanistic reasons to assign one paralog the label "founder" or "parent" (e.g. the template for the RNA in retrogenes or the copy maintaining the ancestral structure in a chimeric duplicate), immediately after duplication, the copies are provisionally assumed to be redundant. And as such, it would only be confusing to label one member the "founding member". The authors even discuss this in relation to the putative redundancy between DAND5 and CER1.
The discussion of cancer on pages 14 and 15 isn't well-integrated into the rest of the manuscript. The reference to RPRM and p53 in particular seems like it could be better incorporated into the narrative of the manuscript. Personally, I'd recommend dropping it, but a smoother integration could also work.
In a manuscript like this one, I would like to see more in depth discussion of sources of error. The task the authors set before themselves is quite ambitious and requires marshaling a lot of data from many genes across many different taxa. These taxa were sequenced by different groups, at different times, with different technology, exhibit different levels of contiguity and likely accuracy and completeness, etc. Sources of error can include errors in multiple alignment, misannotation of the genes, and evolution in gene structure, all of which can lead to aligned non-homologous residues. Moreover, low assembly or annotation completeness can lead to missing genes.
Minor comments
Why use the common name "elephant fish" when there is an "elephant fish" in both Actinopterygii and Chondrichthyes? Perhaps "elephant shark" would be better?
Why didn't the authors use Rhincodon typus (whale shark: https://www.ncbi.nlm.nih.gov/assembly/GCF_001642345.1/) in the analysis? It has a Genbank annotation and appears to be more contiguous than the elephant shark. Also, since this manuscript was posted, there is now a much better Chondrichthyes genome (Pristis pectinata): https://www.ncbi.nlm.nih.gov/assembly/?term=Pristis+pectinata
Perhaps either of these two could be valuable in the analysis.
Typo of DAND5: DADN5
Perhaps a labeled, high-level phylogeny would be useful in orienting the readers. One like Figure 1 in this would be a great service to the reader:
Is Urochordate / Urochordata still in common use?
Reviewed by anonymous reviewer 1, 10 Dec 2019
This is a nice reconstruction of the evolution of a complex gene family, the DAN gene family. The authors show strong supporting evidence for the monophyly of 5 major groups and the inter-group relationships among them. While it is useful to see the information about this gene family all together, the novelty of this study is unclear as the authors often refer to previous literature that shows comparable, albeit partial, results.
Minor comments:
1. the authors should provide more information about the alignments produced (length, % gaps).
2. the authors used an evaluation of likelihood scores to determine convergence of the bayesian phylogenetic reconstruction. Although I generally agree with the authors that this method should produce accurate results, most researchers rely on the estimation of ESS values to determine convergence. It would be useful to know how the ESS values correlate with the number of generations required to reach an asymptotic trend in likelihood scores.
3. At the end of the page with the section entitled "Definition of ancestral gene repertoires" the authors state that the "lack of DAND5 in the elephant fish is an artifact of the current genome assembly". Please provide an explanation for this statement.
4.figure 2 and 3: what is the meaning of the double lines associated to some genes? Also, the grey lines represent intervening genes but no information is provided on how large these intervening sections of DNA may be. Depending on the size, they could be affecting the definition of synteny so more information is necessary to support the conclusions based on synteny.




