
Resolving the clutter of naming “Eve’s” descendants
mtDNA "Nomenclutter" and its Consequences on the Interpretation of Genetic Data
Abstract
Recommendation: posted 23 May 2024, validated 24 May 2024
Günther, T. (2024) Resolving the clutter of naming “Eve’s” descendants. Peer Community in Evolutionary Biology, 100716. https://doi.org/10.24072/pci.evolbiol.100716
Recommendation
Nature is complicated and humans often resort to categorization into simplified groups in order to comprehend and manage complex systems. The human mitochondrial genome and its phylogeny are quite complex. Many of those ~16600 base pairs mutated as humans spread across the planet and the resulting phylogeny can be used to illustrate many different aspects of human history and evolution. But it has too many branches and sub-branches to comprehend, which is why major lineages are considered haplogroups. On the highest level, these haplogroups receive capital letters which are then followed by integers and lowercase letters to designate a more fine-scale structure. This nomenclature even inspired semi-fictional literature, such as Bryan Sykes’ “The Seven Daughters of Eve” [1] from 2001 which includes fictional narratives for each of seven “clan mothers” representing seven major European haplogroups (e.g. Helene representing haplogroup H and Tara representing haplogroup T). But apart from categorizing things, humans also like to make exceptions to rules. For instance, not all haplogroup names consist only of letters and numbers but also special characters. And not everything seems logical or intuitive: the deepest split does not include haplogroup A but the most basal lineage is L0. The main letters also do not represent the same level of the tree structure, Sykes’ Katrine representing haplogroup K should not be considered a “daughter of Eve” but (at best) a granddaughter as K is a sub-haplogroup of U (represented by Ursula). This system and the number of haplogroups have not just reached a point where everything has become incredibly complicated despite supposedly simplifying categories. The inherent arbitrariness can also have serious effects on downstream analysis and the interpretation of results depending on how and on what level the authors of a specific study decide to group their individuals.
This situation of potential biases introduced through the choice of haplogroup groupings is the motivation for the study by Bajić, Schulmann and Nowick who are using the quite fitting term “nomenclutter” in their title [2]. They are raising an important issue in the inconsistencies introduced by the practice of somewhat arbitrary haplotype groupings which varies across studies and has no common standards in place making comparisons between studies virtually impossible. The study shows that the outcome of certain standard analyses and the interpretation of results are very sensitive to the decision on how to group the different haplotypes. This effect is especially pronounced for populations of African ancestry where the haplotype nomenclature would cut the phylogenetic tree at higher levels and the definition of different lineages is generally more coarse than for other populations.
But the authors go beyond pointing out this issue, they also suggest solutions. Instead of grouping sequences by their haplogroup code, one could use “algorithm-based groupings” based on the sequence similarity itself or cutting the phylogenetic tree at a common level of the hierarchy. The analysis of the authors shows that this reduces potential biases substantially. But even such groupings would not be without the influence of the user or researcher’s choices as different parameters have to be set to define the level at which groupings are conducted. The authors propose a neat solution, lifting this issue to be resolved during future updates of the mitochondrial haplogroup nomenclature and the phylogeny. Ideally, the research community could agree on centrally defined haplogroup grouping levels (called “macro-”, “meso-”, and “micro-haplogroups” by the authors) which would all represent different scales of events in human history (from global, continental to local). Classifications like that could be provided through central databases and the classifications could be added to commonly used tools for that purpose. If everyone used these groupings, studies would be a lot more comparable and more fine-scale investigations could still resort to the sequences and the tree itself to avoid all grouping.
The experts who reviewed the study have all highlighted its importance of pointing at a very relevant issue. It will take a community effort to improve practices and the current status of this research area. This study provides an important first step and it should be in everyone’s interest to resolve the “nomenclutter”.
References
1. Sykes B. (2001) The seven daughters of Eve: the science that reveals our genetic ancestry. 1st American ed. New York: Norton.
2. Bajić V, Schulmann VH, Nowick K. (2024) mtDNA “Nomenclutter” and its Consequences on the Interpretation of Genetic Data. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.11.19.567721

The recommender in charge of the evaluation of the article and the reviewers declared that they have no conflict of interest (as defined in the code of conduct of PCI) with the authors or with the content of the article. The authors declared that they comply with the PCI rule of having no financial conflicts of interest in relation to the content of the article.
The authors declare that they have received no specific funding for this study.
Evaluation round #2
DOI or URL of the preprint: https://doi.org/10.1101/2023.11.19.567721
Version of the preprint: 2
Author's Reply, 11 May 2024
Decision by Torsten Günther , posted 22 Apr 2024, validated 22 Apr 2024
, posted 22 Apr 2024, validated 22 Apr 2024
Thank you for your revisions which has been appreciated by all reviewers! Aside from one comment, the manuscript seems close to acceptance. Reviewer #1 is highlighting an additional possibility to learn from the Sars-Cov2 nomenclature which could be included in the discussion of the article.
Reviewed by anonymous reviewer 1, 15 Apr 2024
I would like to thank the authors for the very careful and detailed answers (10 pages) to my open questions, which I found very helpful to clarify the remaining "uncertenties" I had. Great to see that the generated data is all publicly accessible! Just a final comment - regarding the mtHG adress - maybe we can also learn or adapt tools from Sars-Cov-2 research - e.g. https://cov-lineages.org/ or https://www.nature.com/articles/s41564-020-0770-5
As there are no open questions (and those which I had were of minor nature), at least for my part, I can say that everything has been addressed and I therefore endorse the manuscript for publication, hoping it gains wider attention and drives a discussion also for future phylogenetic updates (not necessarily limited to humans mtDNA).
https://doi.org/10.24072/pci.evolbiol.100716.rev21Reviewed by Nicole Huber, 10 Apr 2024
The authors response to my comments was very clear and satisfying for me.
I do not have any further critics or questions that need to be resolved.
Reviewed by Joshua Daniel Rubin, 18 Apr 2024
The authors have adequately addressed all points I raised in the first round of review. I have no more comments. I thank the authors for their valuable work.
https://doi.org/10.24072/pci.evolbiol.100716.rev23Evaluation round #1
DOI or URL of the preprint: https://doi.org/10.1101/2023.11.19.567721
Version of the preprint: 1
Author's Reply, 28 Mar 2024
Decision by Torsten Günther, posted 22 Dec 2023, validated 23 Dec 2023
Your preprint has been seen by three different reviewers. They are all quite positive about your study but have some suggested revisions.
Reviewed by anonymous reviewer 1, 22 Dec 2023
Bajic et al raise an important issue regarding the classification and grouping of mitochondrial DNA (mtDNA) haplotypes in population-based studies of mitochondrial genetic diversity. The authors identify inconsistencies and potential biases introduced by the current practice of using “arbitrary” and non-phylogenetically informed secondary haplogroup groupings, further varying across studies, with no standards in place. As a possible solution the authors propose the implementation of phylogenetically meaningful algorithm-based groupings to define a standardized set of macro-haplogroups, meso-haplogroups, and micro-haplogroups.
The manuscript is very detailed on the background, with good insights on the “evolution of mtDNA nomenclature”. The reasoning is clearly presented and the issues presented graphically. There are minor questions that are unclear to me:
From my understanding there is information missing on how macro, meso and micro-haplogroups can be pre-computed based on the current phylogeny and the ABGs presented within this work, as it is only presented as a concept in the discussion. How feasible is it to pre-compute those based on the current phylogeny?
Also on the use of the ABGs - what is the computational cost - runtime of the two approaches given the 1000G AFR example?
What is the input data needed? Can this approach work with partial control region / or only coding region sequences? Can microarray based mtDNA analysis benefit from such an approach, with often a subset of phylogenetically relevant positions covered for analysis - where one can see very often the use of SC or SCL groupings?
How can we make sure that different studies remain comparable, as the resulting ABGs clusters heavily rely on the population under investigation? This indicates that ABGs alone can not solve the issue and that an additional concept of SNP adresses is needed - is my understanding here correct?
Some minor issues:
Abstract: Line 39 -> the current phylogeny includes more than 5,000 haplogroups - see van Oven 2016.
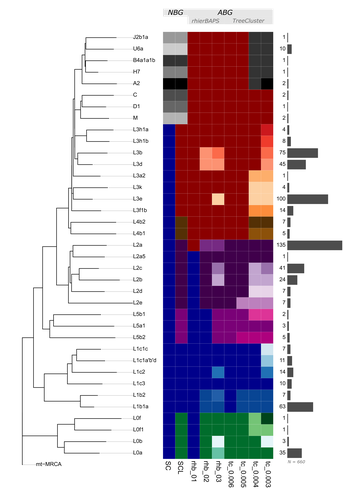
Figure 3 - can you please include the number of clusters for each method - x axis or in the text? Very interesting to see that rhierBAPS clusters L3 and L4 as well as non-african samples (except the U6) together, but also interesting to see from the two-dimensional MDS plots (Figure 2B) that those haplogroups are not further distant.
As the tree method is based on a maximum likelihood (ML) method, whereas the current phylogeny is based on maximum-parsimony (MP) - did you consider a MP-tree clustering approach?
Decreasing the threshold in TreeCluster would be similar to deepen the phylogenetic groupings of the haplogroups on more levels - here my question is - did you consider adding a level to the SCL groups, (e.g. L0a, L0b,..) - that should yield a similar amount of groups as in TreeCluster 0.003 with the advantage that the groups can be estimated without additional clustering.
Figure 4 - for better readability, the methods could be represented in each row (A + B in first, rhierBAPS in row 2 and the TreeCluster Methods in row 3. Did you consider rhierBAPS 04?
Figure 6 - great that the authors point to these issues of the difference between SC and SCL - the question that is however still to answer - is there any of the plots produced by the ABGs that stands out?
It was a bit uncommon to see Results and discussion as one section - Is it possible to split it or does the journal require it this way?
Comments to solutions (line 510 and thereafter)
New Nomenclature: The issue here is that different disciplines take advantage of the mitochondrial phylogeny - as the authors greatly summarized about the introduction of the RSRS , here this work should be included for completeness: https://www.nature.com/articles/jhg2013120
ABGs: Here, the question about intra-study-specific comparison should be addressed. Further one of the main limitations of the ABGs approach presented here I see in the manual curation of the samples to be processed - as described in the methods section.
The concept of the SNP address is a hypothetical one, with several open questions - most prominently how to find the best cutoffs. The most important question is on how many groups to accept for each macro-,miso and micro-haplogroups. Isn’t the concept however rejecting the underlying phylogenetic tree? Can we exclude the possibility of “pseudo-haplogroups” or “polyphyletic clades’ https://www.sciencedirect.com/science/article/pii/S1673852715000405?via%3Dihub
Based on the SNP address it is not possible to find differences between the samples NA19454 and NA1994 on all 3 groupings, or am I missing something?
https://doi.org/10.24072/pci.evolbiol.100716.rev11
Reviewed by Nicole Huber, 04 Dec 2023
Summary
The paper “mtDNA “Nomenclutter” and its Consequences on the Interpretation of Genetic Data” addresses current problems within the nomenclature system of human mitochondrial DNA.
The authors explore the effects of the current nomenclature system to the outcomes of mtDNA research. They describe how bioinformatic based downstream analysis are affected by this issue and how it can lead to inconsistent or misleading interpretation of the same genetic data.
The chosen dataset in this study derives from seven African Ancestry populations highlighting that these lineages are not only underrepresented but also suffer the most from the current limitations.
The authors compare two distinct methods of each, nomenclature-based groupings (NBG) and algorithm-based groupings (ABG):
· NBG
o Single character
o Single character and L
· ABG
o rhierBAPS
o TreeCluster
The authors find that the use of ABG method “TreeCluster” reaches the best accordance with current phylogeny and that this method solves some of the existing problems discussed in the introduction.
As a solution to the addressed problems, the authors suggest the following:
I. use an algorithm-based classification tool to avoid biased haplogroup naming in the future.
II. standardized naming guidelines and the introduction of:
a. “macro-”, “meso-” and “micro-” haplogroups.
III. introduction of a new nomenclature concept (e.g. “mtHg-address”)
Overall Impression
The title of the paper is concise and good. The topic of the paper is indeed a current issue in the field of mtDNA analysis that needs to be addressed and a solution or consensus on the topic would be beneficial for the research community.
The introduction is well written and contains all important parts to introduce the reader to the topic. Literature research was carried out and seems sufficient to me. The used methodology makes sense and is described well.
The data availability section and the authors contribution section are clear and well structured.
Critiques and Improvements
Major Issues
1. I would like to see a more diverse dataset containing not only lineages from African Ancestry. I think it is important to show what effects the proposed method has on non-African data as well since the majority of the data is non-African. An example for haplogroup H would be good to see. I am interested how the fine-resolved haplogroup H would be affected in contrast to the African ancestry lineages.
2. Table S2 is missing.
3. Line 576: The suggestion of 'mt-Hg-address' is interesting. However, a naming system based solely on numbers may not be intuitive for humans. It would be beneficial to explore other naming conventions (e.g., letters, letters & numbers, Continental Regions). For instance, is it possible to create a continental mt-HG-address (e.g., African.1.1.1.1)?
4. Suggestions about the mapping between new and old haplogroup names are missing. How could this be done. Are there examples of the Y-DNA?
Minor Issues
1. Line 39: The current PhyloTree holds over 5,400 nodes and the forensic updated version (as mentioned later in line 131) holds over 6,300 nodes. So I would change the first sentence: “…into more than 2000 described haplogroups,…”.
2. Line 137: besides “macro-haplogroup” and “sub-haplogroup”, there are terms like “meta-haplogroup” and “superhaplogroups” in the current literature. A more detailed overview about existing terms would be valuable. It would also highlight the problem of ambiguous naming but same meaning.
3. The colours in Figure 3 are not clear for me. Maybe add explanation for the colours.
4. Are there alternatives to the ABG presented in this paper? If so, why did the authors choose the methods described here? Are there alternatives to "SNP-address"?
https://doi.org/10.24072/pci.evolbiol.100716.rev12



