
FUMAGALLI Matteo
- School of Biological and Behavioural Sciences, Queen Mary University of London, London, United Kingdom
- Adaptation, Bioinformatics & Computational Biology, Human Evolution, Population Genetics / Genomics
- recommender
Recommendations: 5
Reviews: 0
Recommendations: 5
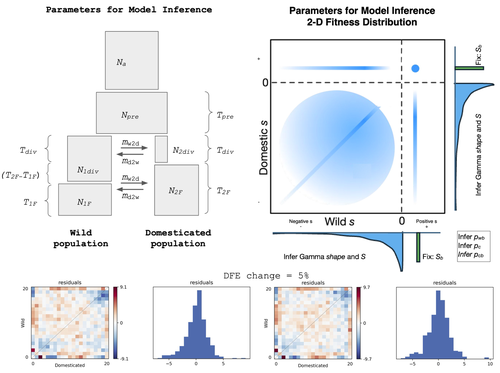
Detection of Domestication Signals through the Analysis of the Full Distribution of Fitness Effects
How the analysis of the distribution of fitness effects can reveal novel insights onto the genetics of domestication
Recommended by Matteo Fumagalli based on reviews by Miguel de Navascués and 1 anonymous reviewerThe joint full distribution of fitness effects (DEF) is an important indicator in population genetic studies, and its inference has been the subject of intense research [1]. However, we still lack a solid framework to estimate DFE under certain demographic conditions.
In this study, Castellano and colleagues propose to estimate the DFE by analysing the site frequency spectrum (SFS), and specifically develop a new approach for the joint DFE model inference [2]. The latter is based on the proportion of variants with divergent selection coefficients. Authors performed extensive simulations under models of domestication which is arguably one of the most crucial series of events in human evolution [3]. Domestication is associated with significant genetic costs in animals [4].
While DFE is typically estimated by contrasting SFS of silent and functional mutations [5], it has been recently suggested to use the joint SFS between domesticated and wild populations to estimate the DFE [6]. Authors build on this model and expand its parameterisation. Authors were able to dissect the impact of linked selection on inferred demographic history of wild and domesticated populations, with a robust estimation of the deleterious DFE.
There are still several limitations in the interpretation of DFE as, for instance, some selective sweeps can bias their estimates and some demographic scenarios are challenging to infer. Also, classic quantitative trait models should be evaluated as a complementary approach. Finally, the in silico predictions presented in this study could be validated by empirical scans on existing genomic data sets. Nevertheless, this study is an important contribution to our understanding on how demography, and domestication in particular, can affect variants under selection in recent evolutionary histories.
References
[1] Eyre-Walker A, Keightley PD. The distribution of fitness effects of new mutations. Nat Rev Genet. 2007;8(8):610-618. https://doi.org/10.1038/nrg2146
[2] Castellano D, Vourlaki IT, Gutenkunst RN, Ramos-Onsins SE. Detection of Domestication Signals through the Analysis of the Full Distribution of Fitness Effects. BioRxiv 2025, ver.4 peer-reviewed and recommended by PCI Evol Biol https://doi.org/10.1101/2022.08.24.505198
[3] Frantz LAF, Bradley DG, Larson G, Orlando L. Animal domestication in the era of ancient genomics. Nat Rev Genet. 2020;21(8):449-460. https://doi.org/10.1038/s41576-020-0225-0
[4] Schubert M, Jónsson H, Chang D, et al. Prehistoric genomes reveal the genetic foundation and cost of horse domestication. Proc Natl Acad Sci U S A. 2014;111(52):E5661-E5669. https://doi.org/10.1073/pnas.1416991111
[5] Kousathanas A, Keightley PD. A comparison of models to infer the distribution of fitness effects of new mutations. Genetics. 2013;193(4):1197-1208. https://doi.org/10.1534/genetics.112.148023
[6] Huang X, Fortier AL, Coffman AJ, et al. Inferring Genome-Wide Correlations of Mutation Fitness Effects between Populations. Mol Biol Evol. 2021;38(10):4588-4602. https://doi.org/10.1093/molbev/msab162

Faster model-based estimation of ancestry proportions
fastmixture generates fast and accurate estimates of global ancestry proportions and ancestral allele frequencies
Recommended by Matteo Fumagalli based on reviews by Oscar Lao Grueso and 2 anonymous reviewersThe estimation of ancestry proportions in individuals is an important analysis in both evolutionary biology and medical genetics. However, popular tools like ADMIXTURE (Alexander et al. 2009) and STRUCTURE (Pritchard et al. 2000) do not scale well with the large amount of data currently available. Recent alternative methods, such as SCOPE (Chiu et al. 2022), favour scalability over accuracy.
In this study, Santander and coworkers introduce a new software, called fastmixture, which estimates ancestry proportions and ancestral allele frequencies using novel implementations for initialisation and convergence of its model-based algorithm (Santander et al. 2024). In simulated datasets, fastmixture displays desirable properties of speed and accuracy, with its performance surpassing commonly used software (Alexander et al. 2009, Pritchard et al. 2000, Chiu et al. 2022, Mantes et al. 2023). fastmixture is almost 30 times faster than ADMIXTURE under a complex model with five ancestral populations, while retaining similar accuracy levels. When applied to data from the 1000 Genomes Project (1000 Genomes Project Consortium 2025), fastmixture recapitulated expected levels of global ancestry. The new software is freely available on GitHub with an accessible documentation. fastmixture accepts input files in PLINK format.
It remains an open question whether extensive parameter tuning could increase the scalability and accuracy of established methods. A comprehensive assessment of fastmixture over a wide range of data processing options (Hemstrom et al. 2024) is also missing. Finally, whether model-based approaches are fully scalable to ever increasing biobank datasets is still under debate. Nevertheless, the superior computational performance of fastmixture is evident and it is likely that this new software will soon replace existing popular tools to estimate global ancestry proportions.
References
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655-1664. https://doi.org/10.1101/gr.094052.109
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945-959. https://doi.org/10.1093/genetics/155.2.945
Chiu AM, Molloy EK, Tan Z, Talwalkar A, Sankararaman S. Inferring population structure in biobank-scale genomic data. Am J Hum Genet. 2022;109(4):727-737. https://doi.org/10.1016/j.ajhg.2022.02.015
Santander CG, Refoyo Martinez A, Meisner J. Faster model-based estimation of ancestry proportions. bioRxiv 2024; ver.2 peer-reviewed and recommended by PCI Evol Biol. https://doi.org/10.1101/2024.07.08.602454
Mantes AD, Montserrat DM, Bustamante CD, Giró-I-Nieto X, Ioannidis AG. Neural ADMIXTURE for rapid genomic clustering. Nat Comput Sci. 2023;3(7):621-629. https://doi.org/10.1038/s43588-023-00482-7
1000 Genomes Project Consortium, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68-74. https://doi.org/10.1038/nature15393
Hemstrom W, Grummer JA, Luikart G, Christie MR. Next-generation data filtering in the genomics era. Nat Rev Genet. 2024;25(11):750-767. https://doi.org/10.1038/s41576-024-00738-6
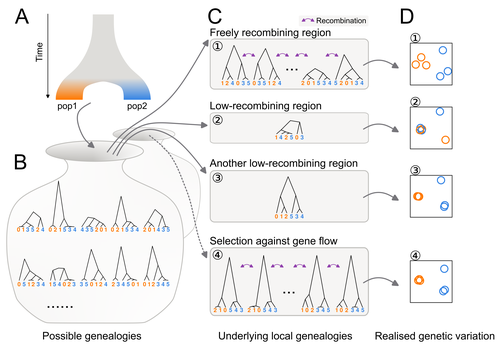
Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structure
Discerning the causes of local deviations in genetic variation: the effect of low-recombination regions
Recommended by Matteo Fumagalli based on reviews by Claire Merot and 1 anonymous reviewerIn this study, Ishigohoka and colleagues tackle an important, yet often overlooked, question on the causes of genetic variation. While genome-wide patterns represent population structure, local variation is often associated with selection. Authors propose that an alternative cause for variation in individual loci is reduced recombination rate.
To test this hypothesis, authors perform local Principal Component Analysis (PCA) (Li & Ralph, 2019) to identify local deviations in population structure in the Eurasian blackcap (Sylvia atricapilla) (Ishigohoka et al. 2022). This approach is typically used to detect chromosomal rearrangements or any long region of linked loci (e.g., due to reduced recombination or selection) (Mérot et al. 2021). While other studies investigated the effect of low recombination on genetic variation (Booker et al. 2020), here authors provide a comprehensive analysis of the effect of recombination to local PCA patterns both in empirical and simulated data sets. Findings demonstrate that low recombination (and not selection) can be the sole explanatory variable for outlier windows. The study also describes patterns of genetic variation along the genome of Eurasian blackcaps, localising at least two polymorphic inversions (Ishigohoka et al. 2022).
Further investigations on the effect of model parameters (e.g., window sizes and thresholds for defining low-recombining regions), as well as the use of powerful neutrality tests are in need to clearly assess whether outlier regions experience selection and reduced recombination, and to what extent.
References
Booker, T. R., Yeaman, S., & Whitlock, M. C. (2020). Variation in recombination rate affects detection of outliers in genome scans under neutrality. Molecular Ecology, 29 (22), 4274–4279. https://doi.org/10.1111/mec.15501
Ishigohoka, J., Bascón-Cardozo, K., Bours, A., Fuß, J., Rhie, A., Mountcastle, J., Haase, B., Chow, W., Collins, J., Howe, K., Uliano-Silva, M., Fedrigo, O., Jarvis, E. D., Pérez-Tris, J., Illera, J. C., Liedvogel, M. (2022) Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structure. bioRxiv 2021.12.22.473882, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.12.22.473882
Li, H., & Ralph, P. (2019). Local PCA Shows How the Effect of Population Structure Differs Along the Genome. Genetics, 211 (1), 289–304. https://doi.org/10.1534/genetics.118.301747
Mérot, C., Berdan, E. L., Cayuela, H., Djambazian, H., Ferchaud, A.-L., Laporte, M., Normandeau, E., Ragoussis, J., Wellenreuther, M., & Bernatchez, L. (2021). Locally Adaptive Inversions Modulate Genetic Variation at Different Geographic Scales in a Seaweed Fly. Molecular Biology and Evolution, 38 (9), 3953–3971. https://doi.org/10.1093/molbev/msab143

Connectivity and selfing drives population genetic structure in a patchy landscape: a comparative approach of four co-occurring freshwater snail species
Determinants of population genetic structure in co-occurring freshwater snails
Recommended by Trine Bilde and Matteo Fumagalli based on reviews by 3 anonymous reviewersGenetic diversity is a key aspect of biodiversity and has important implications for evolutionary potential and thereby the persistence of species. Improving our understanding of the factors that drive genetic structure within and between populations is, therefore, a long-standing goal in evolutionary biology. However, this is a major challenge, because of the complex interplay between genetic drift, migration, and extinction/colonization dynamics on the one hand, and the biology and ecology of species on the other hand (Romiguier et al. 2014, Ellegren and Galtier 2016, Charlesworth 2003).
Jarne et al. (2021) studied whether environmental and demographic factors affect the population genetic structure of four species of hermaphroditic freshwater snails in a similar way, using comparative analyses of neutral genetic microsatellite markers.
Specifically, they investigated microsatellite variability of Hygrophila in almost 280 sites in Guadeloupe, Lesser Antilles, as part of a long-term survey experiment (Lamy et al. 2013). They then modelled the influence of the mating system, local environmental characteristics and demographic factors on population genetic diversity.
Consistent with theoretical predictions (Charlesworth 2003), they detected higher genetic variation in two outcrossing species than in two selfing species, emphasizing the importance of the mating system in maintaining genetic diversity. The study further identified an important role of site connectivity, through its influences on effective population size and extinction/colonisation events. Finally, the study detects an influence of interspecific interactions caused by an ongoing invasion by one of the studied species on genetic structure, highlighting the indirect effect of changes in community composition and demography on population genetics.
Jarne et al. (2021) could address the extent to which genetic structure is determined by demographic and environmental factors in multiple species given the remarkable sampling available. Additionally, the study system is extremely suitable to address this hypothesis as species’ habitats are defined and delineated. Whilst the authors did attempt to test for across-species correlations, further investigations on this matter are required. Moreover, the effect of interactions between factors should be appropriately considered in any modelling between genetic structure and local environmental or demographic features.
The findings in this study contribute to improving our understanding of factors influencing population genetic diversity, and highlights the complexity of interacting factors, therefore also emphasizing the challenges of drawing general implications, additionally hampered by the relatively limited number of species studied. Jarne et al. (2021) provide an excellent showcase of an empirical framework to test determinants of genetic structure in natural populations. As such, this study can be an example for further attempts of comparative analysis of genetic diversity.
References
Charlesworth, D. (2003) Effects of inbreeding on the genetic diversity of populations. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences, 358, 1051-1070. doi: https://doi.org/10.1098/rstb.2003.1296
Ellegren, H. and Galtier, N. (2016) Determinants of genetic diversity. Nature Reviews Genetics, 17, 422-433. doi: https://doi.org/10.1038/nrg.2016.58
Jarne, P., Lozano del Campo, A., Lamy, T., Chapuis, E., Dubart, M., Segard, A., Canard, E., Pointier, J.-P. and David, P. (2021) Connectivity and selfing drives population genetic structure in a patchy landscape: a comparative approach of four co-occurring freshwater snail species. HAL, hal-03295242, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://hal.archives-ouvertes.fr/hal-03295242
Lamy, T., Gimenez, O., Pointier, J. P., Jarne, P. and David, P. (2013). Metapopulation dynamics of species with cryptic life stages. The American Naturalist, 181, 479-491. doi: https://doi.org/10.1086/669676
Romiguier, J., Gayral, P., Ballenghien, M. et al. (2014) Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature, 515, 261-263. doi: https://doi.org/10.1038/nature13685
Power and limits of selection genome scans on temporal data from a selfing population
Detecting loci under natural selection from temporal genomic data of selfing populations
Recommended by Matteo Fumagalli based on reviews by Christian Huber and 2 anonymous reviewersThe observed levels of genomic diversity in contemporary populations are the result of changes imposed by several evolutionary processes. Among them, natural selection is known to dramatically shape the genetic diversity of loci associated with phenotypes which affect the fitness of carriers. As such, many efforts have been dedicated towards developing methods to detect signatures of natural selection from genomes of contemporary samples [1].
Recent technological advances made the generation of large-scale genomic data from temporal samples, either from experimental populations or historical or ancient samples, accessible to a wide scientific community [2]. Notably, temporal population genomic data allow for a direct observation and study of how, for instance, allele frequencies change through time in response to evolutionary stimuli. Such information can be exploited to detect loci under natural selection, either via mathematical modelling or by investigating empirical distributions [3].
However, most of current methods to detect selection from temporal genomic data have largely ignored selfing populations, despite the latter comprising a significant proportion of species with social and economic importance. Selfing changes genomic patterns by reducing the effective recombination rate, which makes distinguishing between neutral evolution and natural selection even more challenging than for the case of outcrossing populations [4]. Nevertheless, an outlier-approach based on temporal genomic data for the selfing Arabidopsis thaliana population revealed loci under selection [5].
This study suggested the promise of detecting selection for selfing populations and encouraged further investigations to test the power of selection scans under different mating systems.
To address this question, Navascués et al. [6] extended a previously proposed approach for temporal genome scan [7] to incorporate partial self-fertilization. In the original implementation [7], it is assumed that, under neutrality, all loci provide levels of genetic differentiation drawn from the same distribution. If some of the loci are under selection, such distribution should show heterogeneity. Navascués et al. [6] proposed a test for the homogeneity between loci-specific and genome-wide differentiation by deriving a null distribution of FST via simulations using SLiM [8]. After filtering for low-frequency variants and correct for multiple tests, authors derived a statistical test for selection and assess its power under a wide range of scenarios of selfing rate, selection coefficient, duration and type of selection [6].
The newly proposed test achieved good performance to distinguish between neutral and selected loci in most tested scenarios.
As expected, the test's performance significantly drops for scenarios of high selfing rates and selection from standing variation. Additionally, the probability to correctly detect selection decreases with increasing distance from the causal variant. Intriguingly, the test showed high power when the selected ancestral allele had an initial low frequency, and when the selected derived allele had a high initial frequency. When applied to a data set of around 1,000 SNPs from the highly selfing Medicago truncatula population, an annual plant of the legume family [9], the test did not provide any candidate loci under selection [6].
In summary, the detection of loci under selection in selfing populations is and largely remains a challenging task even when explictly account for the different mating system. However, recombination events that occurred before the selective pressure allow ancestral beneficial alleles to exhibit a detectable pattern of non-neutrality. As such, in partially selfing populations, the strength of the footprint of selection depends on several factors, mostly on the selfing rate, the time of onset and type of selection.
One major assumption of this study is that the model implies unstructured population and continuity between samples obtained from the same geographical location over time. As such assumptions are typically violated in real populations, further research into the effect of more complex demographic scenarios is desired to fully understand the power to detect selection in selfing populations. Furthermore, more power could be gained by including additional genomic information at each time point. In this context, recent approaches that make full use of genomic data based on deep learning [10] may contribute significantly towards this goal. Similarly, the effect of data filtering on the power to detect selection should be further explored, especially in the context of DNA resequencing experiments. These analyses will help elucidate the power offered by selection scans from temporal genomic data in selfing populations.
References
[1] Stern AJ, Nielsen R (2019) Detecting Natural Selection. In: Handbook of Statistical Genomics , pp. 397–40. John Wiley and Sons, Ltd. https://doi.org/10.1002/9781119487845.ch14
[2] Leonardi M, Librado P, Der Sarkissian C, Schubert M, Alfarhan AH, Alquraishi SA, Al-Rasheid KAS, Gamba C, Willerslev E, Orlando L (2017) Evolutionary Patterns and Processes: Lessons from Ancient DNA. Systematic Biology, 66, e1–e29. https://doi.org/10.1093/sysbio/syw059
[3] Dehasque M, Ávila‐Arcos MC, Díez‐del‐Molino D, Fumagalli M, Guschanski K, Lorenzen ED, Malaspinas A-S, Marques‐Bonet T, Martin MD, Murray GGR, Papadopulos AST, Therkildsen NO, Wegmann D, Dalén L, Foote AD (2020) Inference of natural selection from ancient DNA. Evolution Letters, 4, 94–108. https://doi.org/10.1002/evl3.165
[4] Vitalis R, Couvet D (2001) Two-locus identity probabilities and identity disequilibrium in a partially selfing subdivided population. Genetics Research, 77, 67–81. https://doi.org/10.1017/S0016672300004833
[5] Frachon L, Libourel C, Villoutreix R, Carrère S, Glorieux C, Huard-Chauveau C, Navascués M, Gay L, Vitalis R, Baron E, Amsellem L, Bouchez O, Vidal M, Le Corre V, Roby D, Bergelson J, Roux F (2017) Intermediate degrees of synergistic pleiotropy drive adaptive evolution in ecological time. Nature Ecology and Evolution, 1, 1551–1561. https://doi.org/10.1038/s41559-017-0297-1
[6] Navascués M, Becheler A, Gay L, Ronfort J, Loridon K, Vitalis R (2020) Power and limits of selection genome scans on temporal data from a selfing population. bioRxiv, 2020.05.06.080895, ver. 4 peer-reviewed and recommended by PCI Evol Biol. https://doi.org/10.1101/2020.05.06.080895
[7] Goldringer I, Bataillon T (2004) On the Distribution of Temporal Variations in Allele Frequency: Consequences for the Estimation of Effective Population Size and the Detection of Loci Undergoing Selection. Genetics, 168, 563–568. https://doi.org/10.1534/genetics.103.025908
[8] Messer PW (2013) SLiM: Simulating Evolution with Selection and Linkage. Genetics, 194, 1037–1039. https://doi.org/10.1534/genetics.113.152181
[9] Siol M, Prosperi JM, Bonnin I, Ronfort J (2008) How multilocus genotypic pattern helps to understand the history of selfing populations: a case study in Medicago truncatula. Heredity, 100, 517–525. https://doi.org/10.1038/hdy.2008.5
[10] Sanchez T, Cury J, Charpiat G, Jay F Deep learning for population size history inference: Design, comparison and combination with approximate Bayesian computation. Molecular Ecology Resources, n/a. https://doi.org/10.1111/1755-0998.13224