
Latest recommendations

| Id | Title | Authors | Abstract▲ | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
14 May 2020

Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisonsFrom populations to subspecies… to species? Contrasting patterns of local adaptation in closely-related taxa and their potential contribution to species divergenceRecommended by Emmanuelle Porcher based on reviews by Sophie Karrenberg, Santiago C. Gonzalez-Martinez and 1 anonymous reviewerElevation gradients are convenient and widely used natural setups to study local adaptation, particularly in these times of rapid climate change [e.g. 1]. Marin and her collaborators [2] did not follow the mainstream, however. Instead of tackling adaptation to climate change, they used elevation gradients to address another crucial evolutionary question [3]: could adaptation to altitude lead to ecological speciation, i.e. reproductive isolation between populations in spite of gene flow? More specifically, they examined how much local adaptation to environmental variation differed among closely-related, recently diverged subspecies. They studied several populations of two subspecies of snapdragon (Antirrhinum majus), with adjacent geographical distributions. Using common garden experiments and the classical, but still useful, QST-FST comparison, they demonstrate contrasting patterns of local adaptation to altitude between the two subspecies, with several traits under divergent selection in A. majus striatum but none in A. majus pseudomajus. These differences in local adaptation may contribute to species divergence, and open many stimulating questions on the underlying mechanisms, such as the identity of environmental drivers or contribution of reproductive isolation involving flower color polymorphism. References [1] Anderson, J. T., and Wadgymar, S. M. (2020). Climate change disrupts local adaptation and favours upslope migration. Ecology letters, 23(1), 181-192. doi: 10.1111/ele.13427 | Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisons | Sara Marin, Anaïs Gibert, Juliette Archambeau, Vincent Bonhomme, Mylène Lascoste and Benoit Pujol | <p>Phenotypic divergence among natural populations can be explained by natural selection or by neutral processes such as drift. Many examples in the literature compare putatively neutral (FST) and quantitative genetic (QST) differentiation in mult... | | Adaptation, Evolutionary Ecology, Genotype-Phenotype, Morphological Evolution, Quantitative Genetics | Emmanuelle Porcher | 2018-08-05 15:34:30 | ||
03 Apr 2017
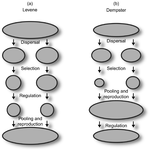
Things softly attained are long retained: Dissecting the Impacts of Selection Regimes on Polymorphism Maintenance in Experimental Spatially Heterogeneous EnvironmentsExperimental test of the conditions of maintenance of polymorphism under hard and soft selectionRecommended by Stephanie Bedhomme based on reviews by Joachim Hermisson and 2 anonymous reviewers
Theoretical work, initiated by Levene (1953) [1] and Dempster (1955) [2], suggests that within a given environment, the way populations are regulated and contribute to the next generation is a key factor for the maintenance of local adaptation polymorphism. In this theoretical context, hard selection describes the situation where the genetic composition of each population affects its contribution to the next generation whereas soft selection describes the case where the contribution of each population is fixed, whatever its genetic composition. Soft selection is able to maintain polymorphism, whereas hard selection invariably leads to the fixation of one of the alleles. Although the specific conditions (e.g. of migration between populations or drift level) in which this prediction holds have been studied in details by theoreticians, experimental tests have mainly failed, usually leading to the conclusion that the allele frequency dynamics was driven by other mechanisms in the experimental systems and conditions used. Gallet, Froissart and Ravigné [3] have set up a bacterial experimental system which allowed them to convincingly demonstrate that soft selection generates the conditions for polymorphism maintenance when hard selection does not, everything else being equal. The key ingredients of their experimental system are (1) the possibility to accurately produce hard and soft selection regimes when daily transferring the populations and (2) the ability to establish artificial well-characterized reproducible trade-offs. To do so, they used two genotypes resisting each one to one antibiotic and combined, across habitats, low antibiotic doses and difference in medium productivity. The experimental approach contains two complementary parts: the first one is looking at changes in the frequencies of two genotypes, initially introduced at around 50% each, over a small number of generations (ca 40) in different environments and selection regimes (soft/hard) and the second one is convincingly showing polymorphism protection by establishing that in soft selection regimes, the lowest fitness genotype is not eliminated even when introduced at low frequency. In this manuscript, a key point is the dialog between theoretical and experimental approaches. The experiments have been thought and designed to be as close as possible to the situations analysed in theoretical work. For example, the experimental polymorphism protection test (experiment 2) closely matches the equivalent analysis classically performed in theoretical approaches. This close fit between theory and experiment is clearly a strength of this study. This said, the experimental system allowing them to realise this close match also has some limitations. For example, changes in allele frequencies could only be monitored over a quite low number of generations because a longer time-scale would have allowed the contribution of de novo mutations and the likely emergence of a generalist genotype resisting to both antibiotics used to generate the local adaptation trade-offs. These limitations, as well as the actual significance of the experimental tests, are discussed in deep details in the manuscript. References [1] Levene H. 1953. Genetic equilibrium when more than one niche is available. American Naturalist 87: 331–333. doi: 10.1086/281792 [2] Dempster ER. 1955. Maintenance of genetic heterogeneity. Cold Spring Harbor Symposia on Quantitative Biology. 20: 25–32. doi: 10.1101/SQB.1955.020.01.005 [3] Gallet R, Froissart R, Ravigné V. 2017. Things softly attained are long retained: dissecting the impacts of selection regimes on polymorphism maintenance in experimental spatially heterogeneous environments. bioRxiv 100743; doi: 10.1101/100743 | Things softly attained are long retained: Dissecting the Impacts of Selection Regimes on Polymorphism Maintenance in Experimental Spatially Heterogeneous Environments | Romain Gallet, Rémy Froissart, Virginie Ravigné | <p>Predicting and managing contemporary adaption requires a proper understanding of the determinants of genetic variation. Spatial heterogeneity of the environment may stably maintain polymorphism when habitat contribution to the next generation c... | | Adaptation, Evolutionary Theory | Stephanie Bedhomme | 2017-01-17 11:06:21 | ||
10 Jan 2019

Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruceDisentangling the recent and ancient demographic history of European spruce speciesRecommended by Jason Holliday based on reviews by 1 anonymous reviewerGenetic diversity in temperate and boreal forests tree species has been strongly affected by late Pleistocene climate oscillations [2,3,5], but also by anthropogenic forces. Particularly in Europe, where a long history of human intervention has re-distributed species and populations, it can be difficult to know if a given forest arose through natural regeneration and gene flow or through some combination of natural and human-mediated processes. This uncertainty can confound inferences of the causes and consequences of standing genetic variation, which may impact our interpretation of demographic events that shaped species before humans became dominant on the landscape. In their paper entitled "Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce", Chen et al. [1] used a genome-wide dataset of 400k SNPs to infer the demographic history of Picea abies (Norway spruce), the most widespread and abundant spruce species in Europe, and to understand its evolutionary relationship with two other spruces (Picea obovata [Siberian spruce] and P. omorika [Serbian spruce]). Three major Norway spruce clusters were identified, corresponding to central Europe, Russia and the Baltics, and Scandinavia, which agrees with previous studies. The density of the SNP data in the present paper enabled inference of previously uncharacterized admixture between these groups, which corresponds to the timing of postglacial recolonization following the last glacial maximum (LGM). This suggests that multiple migration routes gave rise to the extant distribution of the species, and may explain why Chen et al.'s estimates of divergence times among these major Norway spruce groups were older (15mya) than those of previous studies (5-6mya) – those previous studies may have unknowingly included admixed material [4]. Treemix analysis also revealed extensive admixture between Norway and Siberian spruce over the last ~100k years, while the geographically-restricted Serbian spruce was both isolated from introgression and had a dramatically smaller effective population size (Ne) than either of the other two species. This small Ne resulted from a bottleneck associated with the onset of the iron age ~3000 years ago, which suggests that anthropogenic depletion of forest resources has severely impacted this species. Finally, ancestry of Norway spruce samples collected in Sweden and Denmark suggest their recent transfer from more southern areas of the species range. This northward movement of genotypes likely occurred because the trees performed well relative to local provenances, which is a common observation when trees from the south are planted in more northern locations (although at the potential cost of frost damage due to inappropriate phenology). While not the reason for the transfer, the incorporation of southern seed sources into the Swedish breeding and reforestation program may lead to more resilient forests under climate change. Taken together, the data and analysis presented in this paper allowed inference of the intra- and interspecific demographic histories of a tree species group at a very high resolution, and suggest caveats regarding sampling and interpretation of data from areas with a long history of occupancy by humans. References [1] Chen, J., Milesi, P., Jansson, G., Berlin, M., Karlsson, B., Aleksić, J. M., Vendramin, G. G., Lascoux, M. (2018). Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce. BioRxiv, 402016. ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/402016 | Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce | Jun Chen, Lili Li, Pascal Milesi, Gunnar Jansson, Mats Berlin, Bo Karlsson, Jelena Aleksic, Giovanni G Vendramin, Martin Lascoux | <p>Primeval forests are today exceedingly rare in Europe and transfer of forest reproductive material for afforestation and improvement have been very common, especially over the last two centuries. This can be a serious impediment when inferring ... | | Evolutionary Applications, Hybridization / Introgression, Population Genetics / Genomics | Jason Holliday | Anonymous, Anonymous | 2018-08-29 08:33:15 | |
16 Dec 2020

Shifts from pulled to pushed range expansions caused by reduction of landscape connectivityThe push and pull between theory and data in understanding the dynamics of invasionRecommended by Ben Phillips based on reviews by Laura Naslund and 2 anonymous reviewersExciting times are afoot for those of us interested in the ecology and evolution of invasive populations. Recent years have seen evolutionary process woven firmly into our understanding of invasions (Miller et al. 2020). This integration has inspired a welter of empirical and theoretical work. We have moved from field observations and verbal models to replicate experiments and sophisticated mathematical models. Progress has been rapid, and we have seen science at its best; an intimate discussion between theory and data. References Bîrzu, G., Matin, S., Hallatschek, O., and Korolev, K. S. (2019). Genetic drift in range expansions is very sensitive to density dependence in dispersal and growth. Ecology Letters, 22(11), 1817-1827. doi: https://doi.org/10.1111/ele.13364 | Shifts from pulled to pushed range expansions caused by reduction of landscape connectivity | Maxime Dahirel, Aline Bertin, Marjorie Haond, Aurélie Blin, Eric Lombaert, Vincent Calcagno, Simon Fellous, Ludovic Mailleret, Thibaut Malausa, Elodie Vercken | <p>Range expansions are key processes shaping the distribution of species; their ecological and evolutionary dynamics have become especially relevant today, as human influence reshapes ecosystems worldwide. Many attempts to explain and predict ran... | | Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology, Experimental Evolution, Phylogeography & Biogeography | Ben Phillips | 2020-08-04 12:51:56 | ||
05 Dec 2017

Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic dataPredicting small ancestors using contemporary genomes of large mammalsRecommended by Bruce Rannala based on reviews by Bruce Rannala and 1 anonymous reviewerRecent methodological developments and increased genome sequencing efforts have introduced the tantalizing possibility of inferring ancestral phenotypes using DNA from contemporary species. One intriguing application of this idea is to exploit the apparent correlation between substitution rates and body size to infer ancestral species' body sizes using the inferred patterns of substitution rate variation among species lineages based on genomes of extant species [1]. References [1] Romiguier J, Ranwez V, Douzery EJP and Galtier N. 2013. Genomic evidence for large, long-lived ancestors to placental mammals. Molecular Biology and Evolution 30: 5–13. doi: 10.1093/molbev/mss211 [2] Figuet E, Ballenghien M, Lartillot N and Galtier N. 2017. Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic data. bioRxiv, ver. 3 of 4th December 2017. 139147. doi: 10.1101/139147 | Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic data | Emeric Figuet, Marion Ballenghien, Nicolas Lartillot, Nicolas Galtier | <p>Reconstructing ancestral characters on a phylogeny is an arduous task because the observed states at the tips of the tree correspond to a single realization of the underlying evolutionary process. Recently, it was proposed that ancestral traits... | | Genome Evolution, Life History, Macroevolution, Molecular Evolution, Phylogenetics / Phylogenomics | Bruce Rannala | 2017-05-18 15:28:58 | ||
16 Nov 2018

Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen IslandsIntrogression from related species reveals fine-scale structure in an isolated population of mussels and causes patterns of genetic-environment associationsRecommended by Marianne Elias based on reviews by Thomas Broquet and Tatiana GiraudAssessing population connectivity is central to understanding population dynamics, and is therefore of great importance in evolutionary biology and conservation biology. In the marine realm, the apparent absence of physical barriers, large population sizes and high dispersal capacities of most organisms often result in no detectable structure, thereby hindering inferences of population connectivity. In a review paper, Gagnaire et al. [1] propose several ideas to improve detection of population connectivity. Notably, using simulations they show that under certain circumstances introgression from one species into another may reveal cryptic population structure within that second species. References [1] Gagnaire, P.-A., Broquet, T., Aurelle, D., Viard, F., Souissi, A., Bonhomme, F., Arnaud-Haond, S., & Bierne, N. (2015). Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evolutionary Applications, 8, 769–786. doi: 10.1111/eva.12288 | Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands | Christelle Fraïsse, Anne Haguenauer, Karin Gerard, Alexandra Anh-Thu Weber, Nicolas Bierne, Anne Chenuil | <p>Reticulated evolution -i.e. secondary introgression / admixture between sister taxa- is increasingly recognized as playing a key role in structuring infra-specific genetic variation and revealing cryptic genetic connectivity patterns. When admi... | | Hybridization / Introgression, Phylogeography & Biogeography, Population Genetics / Genomics | Marianne Elias | 2017-12-28 14:16:16 | ||
07 Nov 2019

New insights into the population genetics of partially clonal organisms: when seagrass data meet theoretical expectationsInferring rates of clonal versus sexual reproduction from population genetics dataRecommended by Olivier J Hardy based on reviews by Ludwig TRIEST, Stacy Krueger-Hadfield and 1 anonymous reviewerIn partially clonal organisms, genetic markers are often used to characterize the genotypic diversity of populations and infer thereof the relative importance of clonal versus sexual reproduction. Most studies report a measure of genotypic diversity based on a ratio, R, of the number of distinct multilocus genotypes over the sample size, and qualitatively interpret high / low R as indicating the prevalence of sexual / clonal reproduction. However, a theoretical framework allowing to quantify the relative rates of clonal versus sexual reproduction from genotypic diversity is still lacking, except using temporal sampling. Moreover, R is intrinsically highly dependent on sample size and sample design, while alternative measures of genotypic diversity are more robust to sample size, like D*, which is equivalent to the Gini-Simpson diversity index applied to multilocus genotypes. Another potential indicator of reproductive strategies is the inbreeding coefficient, Fis, because population genetics theory predicts that clonal reproduction should lead to negative Fis, at least when the sexual reproduction component occurs through random mating. Taking advantage of this prediction, Arnaud-Haond et al. [1] reanalysed genetic data from 165 populations of four partially clonal seagrass species sampled in a standardized way. They found positive correlations between Fis and both R and D* within each species, reflecting variation in the relative rates of sexual versus clonal reproduction among populations. Moreover, the differences of mean genotypic diversity and Fis values among species were also consistent with their known differences in reproductive strategies. Arnaud-Haond et al. [1] also conclude that previous works based on the interpretation of R generally lead to underestimate the prevalence of clonality in seagrasses. Arnaud-Haond et al. [1] confirm experimentally that Fis merits to be interpreted more properly than usually done when inferring rates of clonal reproduction from population genetics data of species reproducing both sexually and clonally. An advantage of Fis is that it is much less affected by sample size than R, and thus should be more reliable when comparing studies differing in sample design. Hence, when the rate of clonal reproduction becomes significant, we expect Fis < 0 and D* < 1. I expect these two indicators of clonality to be complementary because they rely on different consequences of clonality on pattern of genetic variation. Nevertheless, both measures can be affected by other factors. For example, null alleles, selfing or biparental inbreeding can pull Fis upwards, potentially eliminating the signature of clonal reproduction. Similarly, D* (and other measures of genotypic diversity) can be low because the polymorphism of the genetic markers used is too limited or because sexual reproduction often occurs through selfing, eventually resulting in highly similar homozygous genotypes. References [1] Arnaud-Haond, S., Stoeckel, S., and Bailleul, D. (2019). New insights into the population genetics of partially clonal organisms: when seagrass data meet theoretical expectations. ArXiv:1902.10240 [q-Bio], v6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. Retrieved from http://arxiv.org/abs/1902.10240 | New insights into the population genetics of partially clonal organisms: when seagrass data meet theoretical expectations | Arnaud-Haond, Sophie, Stoeckel, Solenn, and Bailleul, Diane | <p>Seagrass meadows are among the most important coastal ecosystems, in terms of both spatial extent and ecosystem services, but they are also declining worldwide. Understanding the drivers of seagrass meadow dynamics is essential for designing so... | | Evolutionary Ecology, Population Genetics / Genomics, Reproduction and Sex | Olivier J Hardy | 2019-03-01 21:57:34 | ||
06 Jul 2018

Variation in competitive ability with mating system, ploidy and range expansion in four Capsella speciesWhen ecology meets genetics: Towards an integrated understanding of mating system transitions and diversityRecommended by Sylvain Billiard and Henrique Teotonio based on reviews by Yaniv Brandvain, Henrique Teotonio and 1 anonymous reviewerIn the 19th century, C. Darwin and F. Delpino engaged in a debate about the success of species with different reproduction modes, with the later favouring the idea that monoecious plants capable of autonomous selfing could spread more easily than dioecious plants (or self-incompatible hermaphroditic plants) if cross-pollination opportunities were limited [1]. Since then, debate has never faded about how natural selection is responsible for transitions to selfing and can explain the diversity and distribution of reproduction modes we observe in the natural world [2, 3]. References [1] Darwin, C. R. (1876). The effects of cross and self fertilization in the vegetable kingdom. London: Murray.
[2] Stebbins, G. L. (1957). Self fertilization and population variability in the higher plants. The American Naturalist, 91, 337-354. doi: 10.1086/281999 | Variation in competitive ability with mating system, ploidy and range expansion in four Capsella species | Xuyue Yang, Martin Lascoux and Sylvain Glémin | <p>Self-fertilization is often associated with ecological traits corresponding to the ruderal strategy in Grime’s Competitive-Stress-tolerant-Ruderal (CSR) classification of ecological strategies. Consequently, selfers are expected to be less comp... | | Evolutionary Ecology, Population Genetics / Genomics, Reproduction and Sex, Species interactions | Sylvain Billiard | 2017-11-06 19:54:52 | ||
22 Sep 2020
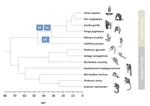
Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primatesStudying genetic antagonisms as drivers of genome evolutionRecommended by Mathieu Joron based on reviews by Qi Zhou and 3 anonymous reviewersSex chromosomes are special in the genome because they are often highly differentiated over much of their lengths and marked by degenerative evolution of their gene content. Understanding why sex chromosomes differentiate requires deciphering the forces driving their recombination patterns. Suppression of recombination may be subject to selection, notably because of functional effects of locking together variation at different traits, as well as longer-term consequences of the inefficient purge of deleterious mutations, both of which may contribute to patterns of differentiation [1]. As an example, male and female functions may reveal intrinsic antagonisms over the optimal genotypes at certain genes or certain combinations of interacting genes. As a result, selection may favour the recruitment of rearrangements blocking recombination and maintaining the association of sex-antagonistic allele combinations with the sex-determining locus. References [1] Charlesworth D (2017) Evolution of recombination rates between sex chromosomes. Philosophical Transactions of the Royal Society B: Biological Sciences, 372, 20160456. https://doi.org/10.1098/rstb.2016.0456 | Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates | Rylan Shearn, Alison E. Wright, Sylvain Mousset, Corinne Régis, Simon Penel, Jean-François Lemaitre, Guillaume Douay, Brigitte Crouau-Roy, Emilie Lecompte, Gabriel A.B. Marais | <p>Sex chromosomes are typically comprised of a non-recombining region and a recombining pseudoautosomal region. Accurately quantifying the relative size of these regions is critical for sex chromosome biology both from a functional (i.e. number o... | | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Reproduction and Sex, Sexual Selection | Mathieu Joron | 2019-02-04 15:16:32 | ||
31 Oct 2022
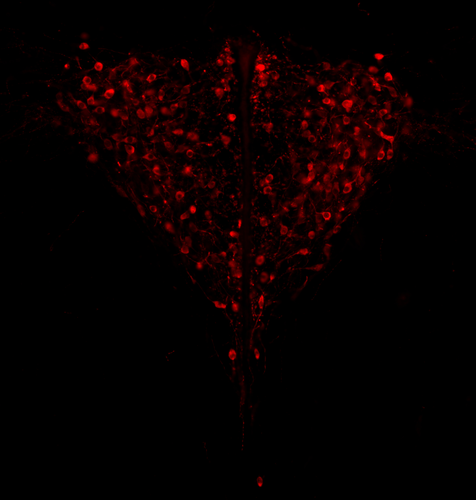
Genotypic sex shapes maternal care in the African Pygmy mouse, Mus minutoidesEffect of sex chromosomes on mammalian behaviour: a case study in pygmy miceRecommended by Gabriel Marais and Trine Bilde based on reviews by Marion Anne-Lise Picard, Caroline Hu and 1 anonymous reviewer based on reviews by Marion Anne-Lise Picard, Caroline Hu and 1 anonymous reviewer
In mammals, it is well documented that sexual dimorphism and in particular sex differences in behaviour are fine-tuned by gonadal hormonal profiles. For example, in lemurs, where female social dominance is common, the level of testosterone in females is unusually high compared to that of other primate females (Petty and Drea 2015). Recent studies however suggest that gonadal hormones might not be the only biological factor involved in establishing sexual dimorphism, sex chromosomes might also play a role. The four core genotype (FCG) model and other similar systems allowing to decouple phenotypic and genotypic sex in mice have provided very convincing evidence of such a role (Gatewood et al. 2006; Arnold and Chen 2009; Arnold 2020a, 2020b). This however is a new field of research and the role of sex chromosomes in establishing sexually dimorphic behaviours has not been studied very much yet. Moreover, the FCG model has some limits. Sry, the male determinant gene on the mammalian Y chromosome might be involved in some sex differences in neuroanatomy, but Sry is always associated with maleness in the FCG model, and this potential role of Sry cannot be studied using this system. Heitzmann et al. have used a natural system to approach these questions. They worked on the African Pygmy mouse, Mus minutoides, in which a modified X chromosome called X* can feminize X*Y individuals, which offers a great opportunity for elegant experiments on the effects of sex chromosomes versus hormones on behaviour. They focused on maternal care and compared pup retrieval, nest quality, and mother-pup interactions in XX, X*X and X*Y females. They found that X*Y females are significantly better at retrieving pups than other females. They are also much more aggressive towards the fathers than other females, preventing paternal care. They build nests of poorer quality but have similar interactions with pups compared to other females. Importantly, no significant differences were found between XX and X*X females for these traits, which points to an effect of the Y chromosome in explaining the differences between X*Y and other females (XX, X*X). Also, another work from the same group showed similar gonadal hormone levels in all the females (Veyrunes et al. 2022). Heitzmann et al. made a number of predictions based on what is known about the neuroanatomy of rodents which might explain such behaviours. Using cytology, they looked for differences in neuron numbers in the hypothalamus involved in the oxytocin, vasopressin and dopaminergic pathways in XX, X*X and X*Y females, but could not find any significant effects. However, this part of their work relied on very small sample sizes and they used virgin females instead of mothers for ethical reasons, which greatly limited the analysis. Interestingly, X*Y females have a higher reproductive performance than XX and X*X ones, which compensate for the cost of producing unviable YY embryos and certainly contribute to maintaining a high frequency of X* in many African pygmy mice populations (Saunders et al. 2014, 2022). X*Y females are probably solitary mothers contrary to other females, and Heitzmann et al. have uncovered a divergent female strategy in this species. Their work points out the role of sex chromosomes in establishing sex differences in behaviours. References Arnold AP (2020a) Sexual differentiation of brain and other tissues: Five questions for the next 50 years. Hormones and Behavior, 120, 104691. https://doi.org/10.1016/j.yhbeh.2020.104691 Arnold AP (2020b) Four Core Genotypes and XY* mouse models: Update on impact on SABV research. Neuroscience & Biobehavioral Reviews, 119, 1–8. https://doi.org/10.1016/j.neubiorev.2020.09.021 Arnold AP, Chen X (2009) What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Frontiers in Neuroendocrinology, 30, 1–9. https://doi.org/10.1016/j.yfrne.2008.11.001 Gatewood JD, Wills A, Shetty S, Xu J, Arnold AP, Burgoyne PS, Rissman EF (2006) Sex Chromosome Complement and Gonadal Sex Influence Aggressive and Parental Behaviors in Mice. Journal of Neuroscience, 26, 2335–2342. https://doi.org/10.1523/JNEUROSCI.3743-05.2006 Heitzmann LD, Challe M, Perez J, Castell L, Galibert E, Martin A, Valjent E, Veyrunes F (2022) Genotypic sex shapes maternal care in the African Pygmy mouse, Mus minutoides. bioRxiv, 2022.04.05.487174, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.04.05.487174 Petty JMA, Drea CM (2015) Female rule in lemurs is ancestral and hormonally mediated. Scientific Reports, 5, 9631. https://doi.org/10.1038/srep09631 Saunders PA, Perez J, Rahmoun M, Ronce O, Crochet P-A, Veyrunes F (2014) Xy Females Do Better Than the Xx in the African Pygmy Mouse, Mus Minutoides. Evolution, 68, 2119–2127. https://doi.org/10.1111/evo.12387 Saunders PA, Perez J, Ronce O, Veyrunes F (2022) Multiple sex chromosome drivers in a mammal with three sex chromosomes. Current Biology, 32, 2001-2010.e3. https://doi.org/10.1016/j.cub.2022.03.029 Veyrunes F, Perez J, Heitzmann L, Saunders PA, Givalois L (2022) Separating the effects of sex hormones and sex chromosomes on behavior in the African pygmy mouse Mus minutoides, a species with XY female sex reversal. bioRxiv, 2022.07.11.499546. https://doi.org/10.1101/2022.07.11.499546 | Genotypic sex shapes maternal care in the African Pygmy mouse, Mus minutoides | Louise D Heitzmann, Marie Challe, Julie Perez, Laia Castell, Evelyne Galibert, Agnes Martin, Emmanuel Valjent, Frederic Veyrunes | <p>Sexually dimorphic behaviours, such as parental care, have long been thought to be driven mostly, if not exclusively, by gonadal hormones. In the past two decades, a few studies have challenged this view, highlighting the direct influence of th... | | Behavior & Social Evolution, Evolutionary Ecology, Reproduction and Sex | Gabriel Marais | 2022-04-08 20:09:58 |




