
Latest recommendations
| Id | Title * | Authors * ▲ | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
10 Jul 2019
Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pestThe scandalous pestRecommended by Nicolas Galtier based on reviews by 2 anonymous reviewers based on reviews by 2 anonymous reviewers
Koutsovoulos et al. [1] have generated and analysed the first population genomic dataset in root-knot nematode Meloidogyne incognita. Why is this interesting? For two major reasons. First, M. incognita has been documented to be apomictic, i.e., to lack any form of sex. This is a trait of major evolutionary importance, with implications on species adaptive potential. The study of genome evolution in asexuals is fascinating and has the potential to inform on the forces governing the evolution of sex and recombination. Even small amounts of sex, however, are sufficient to restore most of the population genetic properties of true sexuals [2]. Because rare events of sex can remain undetected in the field, to confirm asexuality in M. incognita using genomic data is an important step. The second reason why M. incognita is of interest is that this nematode is one of the most harmful pests currently living on earth. M. incognita feeds on the roots of many cultivated plants, including tomato, bean, and cotton, and has been of major agricultural importance for decades. A number of races were defined based on host specificity. These have played a key role in attempts to control the dynamic of M. incognita populations via crop rotations. Races and management strategies so far lack any genetic basis, hence the second major interest of this study. References [1] Koutsovoulos, G. D., Marques, E., Arguel, M. J., Duret, L., Machado, A. C. Z., Carneiro, R. M. D. G., Kozlowski, D. K., Bailly-Bechet, M., Castagnone-Sereno, P., Albuquerque, E. V., & Danchin, E. G. J. (2019). Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pest. bioRxiv, 362129, ver. 5, peer-reviewed and recommended by Peer Community in Evolutionary Biology. doi: 10.1101/362129 | Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pest | Georgios D Koutsovoulos, Eder Marques, Marie-Jeanne Arguel, Laurent Duret, Andressa CZ Machado, Regina MDG Carneiro, Djampa K Kozlowski, Marc Bailly-Bechet, Philippe Castagnone-Sereno, Erika VS Albuquerque, Etienne GJ Danchin | <p>The most devastating nematodes to worldwide agriculture are the root-knot nematodes with Meloidogyne incognita being the most widely distributed and damaging species. This parasitic and ecological success seem surprising given its supposed obli... | | Adaptation, Bioinformatics & Computational Biology, Evolutionary Ecology, Genome Evolution, Genotype-Phenotype, Molecular Evolution, Phylogenetics / Phylogenomics, Population Genetics / Genomics, Reproduction and Sex | Nicolas Galtier | 2018-08-24 09:02:33 | ||
17 May 2021

Relative time constraints improve molecular datingDating with constraintsRecommended by Cécile Ané based on reviews by David Duchêne and 1 anonymous reviewerEstimating the absolute age of diversification events is challenging, because molecular sequences provide timing information in units of substitutions, not years. Additionally, the rate of molecular evolution (in substitutions per year) can vary widely across lineages. Accurate dating of speciation events traditionally relies on non-molecular data. For very fast-evolving organisms such as SARS-CoV-2, for which samples are obtained over a time span, the collection times provide this external information from which we can learn the rate of molecular evolution and date past events (Boni et al. 2020). In groups for which the fossil record is abundant, state-of-the-art dating methods use fossil information to complement molecular data, either in the form of a prior distribution on node ages (Nguyen & Ho 2020), or as data modelled with a fossilization process (Heath et al. 2014). Dating is a challenge in groups that lack fossils or other geological evidence, such as very old lineages and microbial lineages. In these groups, horizontal gene transfer (HGT) events have been identified as informative about relative dates: the ancestor of the gene's donor must be older than the descendants of the gene's recipient. Previous work using HGTs to date phylogenies have used methodologies that are ad-hoc (Davín et al 2018) or employ a small number of HGTs only (Magnabosco et al. 2018, Wolfe & Fournier 2018). Szöllősi et al. (2021) present and validate a Bayesian approach to estimate the age of diversification events based on relative information on these ages, such as implied by HGTs. This approach is flexible because it is modular: constraints on relative node ages can be combined with absolute age information from fossil data, and with any substitution model of molecular evolution, including complex state-of-art models. To ease the computational burden, the authors also introduce a two-step approach, in which the complexity of estimating branch lengths in substitutions per site is decoupled from the complexity of timing the tree with branch lengths in years, accounting for uncertainty in the first step. Currently, one limitation is that the tree topology needs to be known, and another limitation is that constraints need to be certain. Users of this method should be mindful of the latter when hundreds of constraints are used, as done by Szöllősi et al. (2021) to date the trees of Cyanobacteria and Archaea. Szöllősi et al. (2021)'s method is implemented in RevBayes, a highly modular platform for phylogenetic inference, rapidly growing in popularity (Höhna et al. 2016). The RevBayes tutorial page features a step-by-step tutorial "Dating with Relative Constraints", which makes the method highly approachable. References: Boni MF, Lemey P, Jiang X, Lam TT-Y, Perry BW, Castoe TA, Rambaut A, Robertson DL (2020) Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nature Microbiology, 5, 1408–1417. https://doi.org/10.1038/s41564-020-0771-4 Davín AA, Tannier E, Williams TA, Boussau B, Daubin V, Szöllősi GJ (2018) Gene transfers can date the tree of life. Nature Ecology & Evolution, 2, 904–909. https://doi.org/10.1038/s41559-018-0525-3 Heath TA, Huelsenbeck JP, Stadler T (2014) The fossilized birth–death process for coherent calibration of divergence-time estimates. Proceedings of the National Academy of Sciences, 111, E2957–E2966. https://doi.org/10.1073/pnas.1319091111 Höhna S, Landis MJ, Heath TA, Boussau B, Lartillot N, Moore BR, Huelsenbeck JP, Ronquist F (2016) RevBayes: Bayesian Phylogenetic Inference Using Graphical Models and an Interactive Model-Specification Language. Systematic Biology, 65, 726–736. https://doi.org/10.1093/sysbio/syw021 Magnabosco C, Moore KR, Wolfe JM, Fournier GP (2018) Dating phototrophic microbial lineages with reticulate gene histories. Geobiology, 16, 179–189. https://doi.org/10.1111/gbi.12273 Nguyen JMT, Ho SYW (2020) Calibrations from the Fossil Record. In: The Molecular Evolutionary Clock: Theory and Practice (ed Ho SYW), pp. 117–133. Springer International Publishing, Cham. https://doi.org/10.1007/978-3-030-60181-2_8 Szollosi, G.J., Hoehna, S., Williams, T.A., Schrempf, D., Daubin, V., Boussau, B. (2021) Relative time constraints improve molecular dating. bioRxiv, 2020.10.17.343889, ver. 8 recommended and peer-reviewed by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.10.17.343889 Wolfe JM, Fournier GP (2018) Horizontal gene transfer constrains the timing of methanogen evolution. Nature Ecology & Evolution, 2, 897–903. https://doi.org/10.1038/s41559-018-0513-7 | Relative time constraints improve molecular dating | Gergely J Szollosi, Sebastian Hoehna, Tom A Williams, Dominik Schrempf, Vincent Daubin, Bastien Boussau | <p style="text-align: justify;">Dating the tree of life is central to understanding the evolution of life on Earth. Molecular clocks calibrated with fossils represent the state of the art for inferring the ages of major groups. Yet, other informat... | | Bioinformatics & Computational Biology, Genome Evolution, Phylogenetics / Phylogenomics | Cécile Ané | 2020-10-21 23:39:17 | ||
10 Jan 2020

Probabilities of tree topologies with temporal constraints and diversification shiftsFitting diversification models on undated or partially dated treesRecommended by Nicolas Lartillot based on reviews by Amaury Lambert, Dominik Schrempf and 1 anonymous reviewerPhylogenetic trees can be used to extract information about the process of diversification that has generated them. The most common approach to conduct this inference is to rely on a likelihood, defined here as the probability of generating a dated tree T given a diversification model (e.g. a birth-death model), and then use standard maximum likelihood. This idea has been explored extensively in the context of the so-called diversification studies, with many variants for the models and for the questions being asked (diversification rates shifting at certain time points or in the ancestors of particular subclades, trait-dependent diversification rates, etc). References [1] Didier, G. (2020) Probabilities of tree topologies with temporal constraints and diversification shifts. bioRxiv, 376756, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/376756 | Probabilities of tree topologies with temporal constraints and diversification shifts | Gilles Didier | <p>Dating the tree of life is a task far more complicated than only determining the evolutionary relationships between species. It is therefore of interest to develop approaches apt to deal with undated phylogenetic trees. The main result of this ... | | Bioinformatics & Computational Biology, Macroevolution | Nicolas Lartillot | 2019-01-30 11:28:58 | ||
23 Jan 2020

A novel workflow to improve multi-locus genotyping of wildlife species: an experimental set-up with a known model systemImproving the reliability of genotyping of multigene families in non-model organismsRecommended by François Rousset based on reviews by Sebastian Ernesto Ramos-Onsins, Helena Westerdahl and Thomas BigotThe reliability of published scientific papers has been the topic of much recent discussion, notably in the biomedical sciences [1]. Although small sample size is regularly pointed as one of the culprits, big data can also be a concern. The advent of high-throughput sequencing, and the processing of sequence data by opaque bioinformatics workflows, mean that sequences with often high error rates are produced, and that exact but slow analyses are not feasible. References [1] Ioannidis, J. P. A, Greenland, S., Hlatky, M. A., Khoury, M. J., Macleod, M. R., Moher, D., Schulz, K. F. and Tibshirani, R. (2014) Increasing value and reducing waste in research design, conduct, and analysis. The Lancet, 383, 166-175. doi: 10.1016/S0140-6736(13)62227-8 | A novel workflow to improve multi-locus genotyping of wildlife species: an experimental set-up with a known model system | Gillingham, Mark A. F., Montero, B. Karina, Wilhelm, Kerstin, Grudzus, Kara, Sommer, Simone and Santos, Pablo S. C. | <p>Genotyping novel complex multigene systems is particularly challenging in non-model organisms. Target primers frequently amplify simultaneously multiple loci leading to high PCR and sequencing artefacts such as chimeras and allele amplification... | | Bioinformatics & Computational Biology, Evolutionary Ecology, Genome Evolution, Molecular Evolution | François Rousset | Helena Westerdahl, Sebastian Ernesto Ramos-Onsins, Paul J. McMurdie , Arnaud Estoup, Vincent Segura, Jacek Radwan , Torbjørn Rognes , William Stutz , Kevin Vanneste , Thomas Bigot, Jill A. Hollenbach , Wieslaw Babik , Marie-Christin... | 2019-05-15 17:30:44 | |
12 Jul 2017

Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategiesTowards an integrated scenario to understand evolutionary patterns in A. thalianaRecommended by Xavier Picó based on reviews by Rafa Rubio de Casas and Xavier PicóNobody can ignore that a full understanding of evolution requires an integrated approach from both conceptual and methodological viewpoints. Although some life-history traits, e.g. flowering time, have long been receiving more attention than others, in many cases because the former are more workable than the latter, we must acknowledge that our comprehension about how evolution works is strongly biased and limited. In the Arabidopsis community, such an integration is making good progress as an increasing number of research groups worldwide are changing the way in which evolution is put to the test. This manuscript [1] is a good example of that as the authors raise an important issue in evolutionary biology by combining gene expression and flowering time data from different sources. In particular, the authors explore how variation in flowering time, which determines lifespan, and host immunity defenses co-vary, which is interpreted in terms of co-evolution between the two traits. Interestingly, the authors go beyond that pattern by separating lifespan-dependent from lifespan–independent defense genes, and by showing that defense genes with variants known to impact fitness in the field are among the genes whose expression co-varies most strongly with flowering time. Finally, these results are supported by a simple mathematical model indicating that such a relationship can also be expected theoretically. Overall, the readers will find many conceptual and methodological elements of interest in this manuscript. The idea that evolution is better understood under the scope of life history variation is really exciting and challenging, and in my opinion on the right track for disentangling the inherent complexities of evolutionary research. However, only when we face complexity, we also face its costs and burdens. In this particular case, the well-known co-variation between seed dormancy and flowering time is a missing piece, as well as the identification of (variation in) putative selective pressures accounting for the co-evolution between defense mechanisms and life history (seed dormancy vs. flowering time) along environmental gradients. More intellectual, technical and methodological challenges that with no doubt are totally worth it. Reference [1] Glander S, He F, Schmitz G, Witten A, Telschow A, de Meaux J. 2017. Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategies. bioRxiv ver.1 of June 19, 2017. doi: 10.1101/131136 | Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategies | Glander S, He F, Schmitz G, Witten A, Telschow A, de Meaux J | The selective impact of pathogen epidemics on host defenses can be strong but remains transient. By contrast, life-history shifts can durably and continuously modify the balance between costs and benefits of immunity, which arbitrates the evolutio... | | Adaptation, Evolutionary Ecology, Expression Studies, Life History, Phenotypic Plasticity, Quantitative Genetics, Species interactions | Xavier Picó | Sophie Karrenberg, Rafa Rubio de Casas, Xavier Picó | 2017-06-21 10:57:14 | |
18 Aug 2020
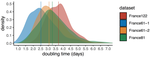
Early phylodynamics analysis of the COVID-19 epidemics in FranceSARS-Cov-2 genome sequence analysis suggests rapid spread followed by epidemic slowdown in FranceRecommended by B. Jesse Shapiro based on reviews by Luca Ferretti and 2 anonymous reviewersSequencing and analyzing SARS-Cov-2 genomes in nearly real time has the potential to quickly confirm (and inform) our knowledge of, and response to, the current pandemic [1,2]. In this manuscript [3], Danesh and colleagues use the earliest set of available SARS-Cov-2 genome sequences available from France to make inferences about the timing of the major epidemic wave, the duration of infections, and the efficacy of lockdown measures. Their phylodynamic estimates -- based on fitting genomic data to molecular clock and transmission models -- are reassuringly close to estimates based on 'traditional' epidemiological methods: the French epidemic likely began in mid-January or early February 2020, and spread relatively rapidly (doubling every 3-5 days), with people remaining infectious for a median of 5 days [4,5]. These transmission parameters are broadly in line with estimates from China [6,7], but are currently unknown in France (in the absence of contact tracing data). By estimating the temporal reproductive number (Rt), the authors detected a slowing down of the epidemic in the most recent period of the study, after mid-March, supporting the efficacy of lockdown measures. References [1] Grubaugh, N. D., Ladner, J. T., Lemey, P., Pybus, O. G., Rambaut, A., Holmes, E. C., & Andersen, K. G. (2019). Tracking virus outbreaks in the twenty-first century. Nature microbiology, 4(1), 10-19. doi: 10.1038/s41564-018-0296-2 | Early phylodynamics analysis of the COVID-19 epidemics in France | Gonché Danesh, Baptiste Elie,Yannis Michalakis, Mircea T. Sofonea, Antonin Bal, Sylvie Behillil, Grégory Destras, David Boutolleau, Sonia Burrel, Anne-Geneviève Marcelin, Jean-Christophe Plantier, Vincent Thibault, Etienne Simon-Loriere, Sylvie va... | <p>France was one of the first countries to be reached by the COVID-19 pandemic. Here, we analyse 196 SARS-Cov-2 genomes collected between Jan 24 and Mar 24 2020, and perform a phylodynamics analysis. In particular, we analyse the doubling time, r... | | Evolutionary Epidemiology, Molecular Evolution, Phylogenetics / Phylogenomics | B. Jesse Shapiro | 2020-06-04 13:13:57 | ||
11 Dec 2020

Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence dataPhylodynamics of hepatitis C virus reveals transmission dynamics within and between risk groups in LyonRecommended by David Rasmussen based on reviews by Chris Wymant and Louis DuPlessisGenomic epidemiology seeks to better understand the transmission dynamics of infectious pathogens using molecular sequence data. Phylodynamic methods have given genomic epidemiology new power to track the transmission dynamics of pathogens by combining phylogenetic analyses with epidemiological modeling. In recent year, applications of phylodynamics to chronic viral infections such as HIV and hepatitis C virus (HVC) have provided some of the best examples of how phylodynamic inference can provide valuable insights into transmission dynamics within and between different subpopulations or risk groups, allowing for more targeted interventions. References [1] Rasmussen, D. A., Volz, E. M., and Koelle, K. (2014). Phylodynamic inference for structured epidemiological models. PLoS Comput Biol, 10(4), e1003570. doi: https://doi.org/10.1371/journal.pcbi.1003570 | Quantifying transmission dynamics of acute hepatitis C virus infections in a heterogeneous population using sequence data | Gonche Danesh, Victor Virlogeux, Christophe Ramière, Caroline Charre, Laurent Cotte, Samuel Alizon | <p>Opioid substitution and syringes exchange programs have drastically reduced hepatitis C virus (HCV) spread in France but HCV sexual transmission in men having sex with men (MSM) has recently arisen as a significant public health concern. The fa... | | Evolutionary Epidemiology, Phylogenetics / Phylogenomics | David Rasmussen | 2019-07-11 13:37:23 | ||
05 Feb 2019
The quiescent X, the replicative Y and the AutosomesReplication-independent mutations: a universal signature ?Recommended by Nicolas Galtier based on reviews by Marc Robinson-Rechavi and Robert Lanfear
Mutations are the primary source of genetic variation, and there is an obvious interest in characterizing and understanding the processes by which they appear. One particularly important question is the relative abundance, and nature, of replication-dependent and replication-independent mutations - the former arise as cells replicate due to DNA polymerization errors, whereas the latter are unrelated to the cell cycle. A recent experimental study in fission yeast identified a signature of mutations in quiescent (=non-replicating) cells: the spectrum of such mutations is characterized by an enrichment in insertions and deletions (indels) compared to point mutations, and an enrichment of deletions compared to insertions [2]. References [1] Achaz, G., Gangloff, S., and Arcangioli, B. (2019). The quiescent X, the replicative Y and the Autosomes. BioRxiv, 351288, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/351288 | The quiescent X, the replicative Y and the Autosomes | Guillaume Achaz, Serge Gangloff, Benoit Arcangioli | <p>From the analysis of the mutation spectrum in the 2,504 sequenced human genomes from the 1000 genomes project (phase 3), we show that sexual chromosomes (X and Y) exhibit a different proportion of indel mutations than autosomes (A), ranking the... | | Bioinformatics & Computational Biology, Genome Evolution, Human Evolution, Molecular Evolution, Population Genetics / Genomics, Reproduction and Sex | Nicolas Galtier | 2018-07-25 10:37:48 | ||
12 Feb 2024
How do plant RNA viruses overcome the negative effect of Muller s ratchet despite strong transmission bottlenecks?How to survive the mutational meltdown: lessons from plant RNA virusesRecommended by Kavita Jain based on reviews by Brent Allman, Ana Morales-Arce and 1 anonymous reviewerAlthough most mutations are deleterious, the strongly deleterious ones do not spread in a very large population as their chance of fixation is very small. Another mechanism via which the deleterious mutations can be eliminated is via recombination or sexual reproduction. However, in a finite asexual population, the subpopulation without any deleterious mutation will eventually acquire a deleterious mutation resulting in the reduction of the population size or in other words, an increase in the genetic drift. This, in turn, will lead the population to acquire deleterious mutations at a faster rate eventually leading to a mutational meltdown. This irreversible (or, at least over some long time scales) accumulation of deleterious mutations is especially relevant to RNA viruses due to their high mutation rate, and while the prior work has dealt with bacteriophages and RNA viruses, the study by Lafforgue et al. [1] makes an interesting contribution to the existing literature by focusing on plants. In this study, the authors enquire how despite the repeated increase in the strength of genetic drift, how the RNA viruses manage to survive in plants. Following a series of experiments and some numerical simulations, the authors find that as expected, after severe bottlenecks, the fitness of the population decreases significantly. But if the bottlenecks are followed by population expansion, the Muller’s ratchet can be halted due to the genetic diversity generated during population growth. They hypothesize this mechanism as a potential way by which the RNA viruses can survive the mutational meltdown. As a theoretician, I find this investigation quite interesting and would like to see more studies addressing, e.g., the minimum population growth rate required to counter the potential extinction for a given bottleneck size and deleterious mutation rate. Of course, it would be interesting to see in future work if the hypothesis in this article can be tested in natural populations. References [1] Guillaume Lafforgue, Marie Lefebvre, Thierry Michon, Santiago F. Elena (2024) How do plant RNA viruses overcome the negative effect of Muller s ratchet despite strong transmission bottlenecks? bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community In Evolutionary Biology | How do plant RNA viruses overcome the negative effect of Muller s ratchet despite strong transmission bottlenecks? | Guillaume Lafforgue, Marie Lefebvre, Thierry Michon, Santiago F. Elena | <p>Muller's ratchet refers to the irreversible accumulation of deleterious mutations in small populations, resulting in a decline in overall fitness. This phenomenon has been extensively observed in experiments involving microorganisms, including ... | | Experimental Evolution, Genome Evolution | Kavita Jain | 2023-08-04 09:37:08 | ||
13 Apr 2023
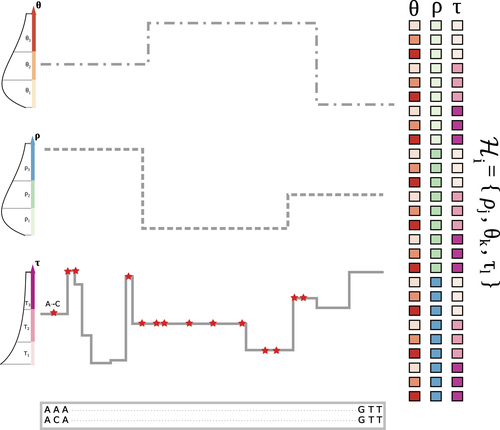
The landscape of nucleotide diversity in Drosophila melanogaster is shaped by mutation rate variationAn unusual suspect: the mutation landscape as a determinant of local variation in nucleotide diversityRecommended by Fernando Racimo based on reviews by David Castellano and 1 anonymous reviewerSometimes, important factors for explaining biological processes fall through the cracks, and it is only through careful modeling that their importance eventually comes out to light. In this study, Barroso and Dutheil introduce a new method based on the sequentially Markovian coalescent (SMC, Marjoran and Wall 2006) for jointly estimating local recombination and coalescent rates along a genome. Unlike previous SMC-based methods, however, their method can also co-estimate local patterns of variation in mutation rates. This is a powerful improvement which allows them to tackle questions about the reasons for the extensive variation in nucleotide diversity across the chromosomes of a species - a problem that has plagued the minds of population geneticists for decades (Begun and Aquadro 1992, Andolfatto 2007, McVicker et al., 2009, Pouyet and Gilbert 2021). The authors find that variation in de novo mutation rates appears to be the most important factor in determining nucleotide diversity in Drosophila melanogaster. Though seemingly contradicting previous attempts at addressing this problem (Comeron 2014), they take care to investigate and explain why that might be the case. Barroso and Dutheil have also taken care to carefully explain the details of their new approach and have carried a very thorough set of analyses comparing competing explanations for patterns of nucleotide variation via causal modeling. The reviewers raised several issues involving choices made by the authors in their analysis of variance partitioning, the proper evaluation of the role of linked selection and the recombination rate estimates emerging from their model. These issues have all been extensively addressed by the authors, and their conclusions seem to remain robust. The study illustrates why the mutation landscape should not be ignored as an important determinant of local variation in genetic diversity, and opens up questions about the generalizability of these results to other organisms. REFERENCES Andolfatto, P. (2007). Hitchhiking effects of recurrent beneficial amino acid substitutions in the Drosophila melanogaster genome. Genome research, 17(12), 1755-1762. https://doi.org/10.1101/gr.6691007 Barroso, G. V., & Dutheil, J. Y. (2021). The landscape of nucleotide diversity in Drosophila melanogaster is shaped by mutation rate variation. bioRxiv, 2021.09.16.460667, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.09.16.460667 Begun, D. J., & Aquadro, C. F. (1992). Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature, 356(6369), 519-520. https://doi.org/10.1038/356519a0 Comeron, J. M. (2014). Background selection as baseline for nucleotide variation across the Drosophila genome. PLoS Genetics, 10(6), e1004434. https://doi.org/10.1371/journal.pgen.1004434 Marjoram, P., & Wall, J. D. (2006). Fast" coalescent" simulation. BMC genetics, 7, 1-9. https://doi.org/10.1186/1471-2156-7-16 McVicker, G., Gordon, D., Davis, C., & Green, P. (2009). Widespread genomic signatures of natural selection in hominid evolution. PLoS genetics, 5(5), e1000471. https://doi.org/10.1371/journal.pgen.1000471 Pouyet, F., & Gilbert, K. J. (2021). Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between. Peer Community Journal, 1, e27. https://doi.org/10.24072/pcjournal.16 | The landscape of nucleotide diversity in Drosophila melanogaster is shaped by mutation rate variation | Gustavo V Barroso, Julien Y Dutheil | <p style="text-align: justify;">What shapes the distribution of nucleotide diversity along the genome? Attempts to answer this question have sparked debate about the roles of neutral stochastic processes and natural selection in molecular evolutio... | | Bioinformatics & Computational Biology, Population Genetics / Genomics | Fernando Racimo | 2022-10-30 07:52:07 |




