
Latest recommendations

| Id | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender▲ | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
05 Dec 2017

Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic dataPredicting small ancestors using contemporary genomes of large mammalsRecommended by Bruce Rannala based on reviews by Bruce Rannala and 1 anonymous reviewerRecent methodological developments and increased genome sequencing efforts have introduced the tantalizing possibility of inferring ancestral phenotypes using DNA from contemporary species. One intriguing application of this idea is to exploit the apparent correlation between substitution rates and body size to infer ancestral species' body sizes using the inferred patterns of substitution rate variation among species lineages based on genomes of extant species [1]. References [1] Romiguier J, Ranwez V, Douzery EJP and Galtier N. 2013. Genomic evidence for large, long-lived ancestors to placental mammals. Molecular Biology and Evolution 30: 5–13. doi: 10.1093/molbev/mss211 [2] Figuet E, Ballenghien M, Lartillot N and Galtier N. 2017. Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic data. bioRxiv, ver. 3 of 4th December 2017. 139147. doi: 10.1101/139147 | Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic data | Emeric Figuet, Marion Ballenghien, Nicolas Lartillot, Nicolas Galtier | <p>Reconstructing ancestral characters on a phylogeny is an arduous task because the observed states at the tips of the tree correspond to a single realization of the underlying evolutionary process. Recently, it was proposed that ancestral traits... | | Genome Evolution, Life History, Macroevolution, Molecular Evolution, Phylogenetics / Phylogenomics | Bruce Rannala | 2017-05-18 15:28:58 | ||
28 Mar 2024
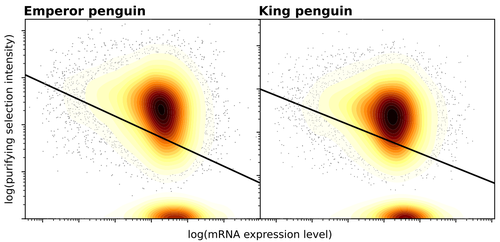
Gene expression is the main driver of purifying selection in large penguin populationsPurifying selection on highly expressed genes in PenguinsRecommended by Bruce Rannala based on reviews by Tanja Pyhäjärvi and 1 anonymous reviewerGiven the general importance of protein expression levels, in cells it is widely accepted that gene expression levels are often a target of natural selection and that most mutations affecting gene expression levels are therefore likely to be deleterious [1]. However, it is perhaps less obvious that the strength of selection on the regulated genes themselves may be influenced by their expression levels. This might be due to harmful effects of misfolded proteins, for example, when higher protein concentrations exist in cells [2]. Recent studies have suggested that highly expressed genes accumulate fewer deleterious mutations; thus a positive relationship appears to exist between gene expression levels and the relative strength of purifying selection [3]. The recommended paper by Trucchi et al. [4] examines the relationship between gene expression, purifying selection and a third variable -- effective population size -- in populations of two species of penguin with different population sizes, the Emperor penguin (Aptenodytes forsteri) and the King penguin (A. patagonicus). Using transcriptomic data and computer simulations modeling selection, they examine patterns of nonsynonymous and synonymous segregating polymorphisms (p) across genes in the two populations, concluding that even in relatively small populations purifying selection has an important effect in eliminating deleterious mutations. References 1] Gilad Y, Oshlack A, and Rifkin SA. 2006. Natural selection on gene expression. Trends in Genetics 22: 456-461. https://doi.org/10.1016/j.tig.2006.06.002 [4] Trucchi E, Massa P, Giannelli F, Latrille T, Fernandes FAN, Ancona L, Stenseth NC, Obiol JF, Paris J, Bertorelle G, and Le Bohec, C. 2023. Gene expression is the main driver of purifying selection in large penguin populations. bioRxiv 2023.08.08.552445, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2023.08.08.552445
| Gene expression is the main driver of purifying selection in large penguin populations | Emiliano Trucchi, Piergiorgio Massa, Francesco Giannelli, Thibault Latrille, Flavia A.N. Fernandes, Lorena Ancona, Nils Chr Stenseth, Joan Ferrer Obiol, Josephine Paris, Giorgio Bertorelle, Celine Le Bohec | <p style="text-align: justify;">Purifying selection is the most pervasive type of selection, as it constantly removes deleterious mutations arising in populations, directly scaling with population size. Highly expressed genes appear to accumulate ... | | Bioinformatics & Computational Biology, Evolutionary Dynamics, Evolutionary Theory, Population Genetics / Genomics | Bruce Rannala | 2023-08-09 17:53:03 | ||
18 Jan 2017

POSTPRINT
Associative Mechanisms Allow for Social Learning and Cultural Transmission of String Pulling in an InsectCulture in BumblebeesRecommended by Caroline Nieberding and Jacques J. M. van AlphenThis is an original paper [1] addressing the question whether cultural transmission occurs in insects and studying the mechanisms of such transmission. Often, culture-like phenomena require relatively sophisticated learning mechanisms, for example imitation and/or teaching. In insects, seemingly complex processes of social information acquisition, can sometimes instead be mediated by relatively simple learning mechanisms suggesting that cultural processes may not necessarily require sophisticated learning abilities. An important quality of this paper is to describe neatly the experimental protocols used for such typically complex behavioural analyses, providing a detailed understanding of the results while it remains a joy to read. This becomes rare in high impact journals. In a clever experimental design, individual bumblebees are trained to pull an artificial flower from under a Plexiglas table to get access to a reward, by pulling a string attached to the flower. Individuals that have learnt this task are then shown to inexperienced bees while performing this task. This results in a large proportion of the inexperienced observers learning to pull the string and getting access to the reward. Finally, the authors could then document the spread of the string pulling skill amongst other workers in the colony. Even when the originally trained individuals had died, the skill of string-pulling persisted in the colony, as long as they were challenged with the task. This shows that cultural transmission takes place within a colony. The authors provide evidence that the transmission of this behavior among individuals relies on a mix of social learning by local enhancement (bees were attracted to the location where they had observed a demonstrator) and of non-social, individual learning (pulling the string is learned by trial and errors and not by direct imitation of the conspecific). Data also show that simple associative mechanisms are enough and that stimulus enhancement was involved (bees were attracted to the string when its location was concordant with that during prior observation). The cleverly designed experiments use a paradigm (string-pulling) which has often been used to investigate cognitive abilities in vertebrates. Comparison with such studies indicate that bees, in some aspects of their learning, may not be different from birds, dogs, or apes as they also relied on the perceptual feedback provided by their actions, resulting in target movement to learn string pulling. The results of the study suggest that the combination of relatively simple forms of social learning and trial-and-error learning can mediate the acquisition of new skills and that bumblebees possess the essential cognitive elements for cultural transmission and in a broader sense, that the capacity of culture may be present within most animals. Can we expect behavioural innovation such as string pulling to occur in nature? Bombus terrestris colonies can reach a total of several hundreds foragers. In the experiments, foragers needed on average 5 rounds of observations with different demonstrators to learn how to pull the string. As individuals forage in a meadow full of flowers and conspecifics, transmission of behavioural innovations by repeated observations shouldn’t strike us as something impossible. Would the behavior survive through the winter? Bumblebee colonies are seasonal in northern areas and in the Mediterranean area but tropical species persists for several years. In seasonal species, all the workers die before winter and only new queens overwinter. So there is no possibility for seasonal foragers to transmit the technique overwinter. Only queens could potentially transmit it to new foragers in spring. However flowers are different in autumn and spring. Therefore, what queens have learnt about flowers in autumn would unlikely be useful in spring (providing that they can remember it). However there is no reason why the technique couldn't be transmitted from a colony to another between spring to autumn. Such transmission of new behaviour would more easily persist in perennial social insect colonies, like honeybees. Importantly, the bees used in these experiments came from a company whose rearing conditions are unknown, and only a few colonies were used for each experiment. As learning ability has a genetic basis [2-3], colonies differ in their ability to learn [4]. In this regard, the authors showed variation between individual bumblebees and between bumblebee colonies in learning ability. Hence, we would wish to know more about the level of genetic diversity in the wild, and of genetic differentiation between tested colonies (were they independent replicates?), to extrapolate the results to what may happen in the wild. Excitingly, the authors found 2 true innovators among the >400 individuals that were tested at least once for 5 min who would solve such a task without stepwise training or observation of skilled demonstrators, showing that behavioural innovation can occur in very small numbers of individuals, provided that an ecological trigger is provided (food reward). Hence this study shows that all ingredients for the long proposed “social heredity” theory proposed by Baldwin in 1896 are available in this organism, suggesting that social transmission of behavioural innovations could technically act as an additional mechanism for adaptive evolution [5], next to genetic evolution that may take longer to produce adaptive evolution. The question remains whether the behavioural innovations are arising from standing genetic variation in the bees, or do not need a firm genetic background to appear. References [1] Alem S, Perry CJ, Zhu X, Loukola OJ, Ingraham T, Søvik E, Chittka L. 2016. Associative mechanisms allow for social learning and cultural transmission of string pulling in an insect. PloS Biology 14:e1002564. doi: 10.1371/journal.pbio.1002564 [2] Mery F, Kawecki TJ. 2002. Experimental evolution of learning ability in fruit flies. Proceeding of the National Academy of Science USA 99:14274-14279. doi: 10.1073/pnas.222371199 [3] Mery F, Belay AT, So AKC, Sokolowski MB, Kawecki TJ. 2007. Natural polymorphism affecting learning and memory in Drosophila. Proceeding of the National Academy of Science USA 104:13051-13055. doi: 10.1073/pnas.0702923104 [4] Raine NE, Chittka L. 2008. The correlation of learning speed and natural foraging success in bumble-bees. Proceeding of the Royal Society of London 275: 803-808. doi : 10.1098/rspb.2007.1652 [5] Baldwin JM. 1896. A New Factor in Evolution. American Naturalist 30:441-451 and 536-553. doi: 10.1086/276408 | Associative Mechanisms Allow for Social Learning and Cultural Transmission of String Pulling in an Insect | Alem S, Perry CJ, Zhu X, Loukola OJ, Ingraham T, Søvik E, Chittka L | Social insects make elaborate use of simple mechanisms to achieve seemingly complex behavior and may thus provide a unique resource to discover the basic cognitive elements required for culture, i.e., group-specific behaviors that spread from “inn... | | Behavior & Social Evolution, Evolutionary Ecology, Non Genetic Inheritance, Phenotypic Plasticity | Caroline Nieberding | 2017-01-18 10:49:03 | ||
28 Feb 2018

Insects and incest: sib-mating tolerance in natural populations of a parasitoid waspIncestuous insects in nature despite occasional fitness costsRecommended by Caroline Nieberding and Bertanne Visser based on reviews by 2 anonymous reviewersInbreeding, or mating between relatives, generally lowers fitness [1]. Mating between genetically similar individuals can result in higher levels of homozygosity and consequently a higher frequency with which recessive disease alleles may be expressed within a population. Reduced fitness as a consequence of inbreeding, or inbreeding depression, can vary between individuals, sexes, populations and species [2], but remains a pervasive challenge for many organisms with small local population sizes, including humans [3]. But all is not lost for individuals within small populations, because an array of mechanisms can be employed to evade the negative effects of inbreeding [4], including sib-mating avoidance and dispersal [5, 6]. Despite thorough investigation of inbreeding and sib-mating avoidance in the laboratory, only very few studies have ventured into the field besides studies on vertebrates and eusocial insects. The study of Collet et al. [7] is a surprising exception, where the effect of male density and frequency of relatives on inbreeding avoidance was tested in the laboratory, after which robust field collections and microsatellite genotyping were used to infer relatedness and dispersal in natural populations. The parasitic wasp Venturia canescens is an excellent model system to study inbreeding, because mating success was previously found to decrease with increasing relatedness between mates in the laboratory [8] and this species thus suffers from inbreeding depression [9–11]. The authors used an elegant design combining population genetics and model simulations to estimate relatedness of mating partners in the field and compared that with a theoretical distribution of potential mate encounters when random mating is assumed. One of the most important findings of this study is that mating between siblings is not avoided in this species in the wild, despite negative fitness effects when inbreeding does occur. Similar findings were obtained for another insect species, the field cricket Gryllus campestris [12], which leaves us to wonder whether inbreeding tolerance could be more common in nature than currently appreciated. The authors further looked into sex-specific dispersal patterns between two patches located a few hundred meters apart. Females were indeed shown to be more related within a patch, but no genetic differences were observed between males, suggesting that V. canescens males more readily disperse. Moreover, microsatellite data at 18 different loci did not reveal genetic differentiation between populations approximately 300 kilometers apart. Gene flow is thus occurring over considerable distances, which could play an important role in the ability of this species to avoid negative fitness consequences of inbreeding in nature. Another interesting aspect of this work is that discrepancies were found between laboratory- and field-based data. What is the relevance of laboratory-based experiments if they cannot predict what is happening in the wild? Many, if not most, biologists (including us) bring our model system into the laboratory to control, at least to some extent, the plethora of environmental factors that could potentially affect our system (in ways that we do not want). Most behavioral studies on mating patterns and sexual selection are conducted in standardized laboratory conditions, but sexual selection is in essence social selection, because an individual’s fitness is partly determined by the phenotype of its social partners (i.e. the social environment) [13]. The social environment may actually dictate the expression of female mate choice and it is unclear how potential laboratory-induced social biases affect mating outcome. In V. canescens, findings using field-caught individuals paint a completely opposite picture of what was previously shown in the laboratory, i.e. sib-avoidance is not taking place in the field. It is likely that density, level of relatedness, sex ratio in the field, and/or the size of experimental arenas in the lab are all factors affecting mate selectivity, as we have previously shown in a butterfly [14–16]. If females, for example, typically only encounter a few males in sequence in the wild, it may be problematic for them to express choosiness when confronted simultaneously with two or more males in the laboratory. A recent study showed that, in the wild, female moths take advantage of staying in groups to blur male choosiness [17]. It is becoming more and more clear that what we observe in the laboratory may not actually reflect what is happening in nature [18]. Instead of ignoring the species-specific life history and ecological features of our favorite species when conducting lab experiments, we suggest that it is time to accept that we now have the theoretical foundations to tease apart what in this “environmental noise” actually shapes sexual selection in nature. Explicitly including ecology in studies on sexual selection will allow us to make more meaningful conclusions, i.e. rather than “this is what may happen in the wild”, we would be able to state “this is what often happens in nature”. References [1] Charlesworth D & Willis JH. 2009. The genetics of inbreeding depression. Nat. Rev. Genet. 10: 783–796. doi: 10.1038/nrg2664 | Insects and incest: sib-mating tolerance in natural populations of a parasitoid wasp | Marie Collet, Isabelle Amat, Sandrine Sauzet, Alexandra Auguste, Xavier Fauvergue, Laurence Mouton, Emmanuel Desouhant | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (http://dx.doi.org/10.24072/pci.evolbiol.100047) 1. Sib-mating avoidance is a pervasive behaviour that likely evolves in species subject to inbreeding dep... | | Behavior & Social Evolution, Evolutionary Ecology, Sexual Selection | Caroline Nieberding | 2017-07-28 09:23:20 | ||
17 May 2021

Relative time constraints improve molecular datingDating with constraintsRecommended by Cécile Ané based on reviews by David Duchêne and 1 anonymous reviewerEstimating the absolute age of diversification events is challenging, because molecular sequences provide timing information in units of substitutions, not years. Additionally, the rate of molecular evolution (in substitutions per year) can vary widely across lineages. Accurate dating of speciation events traditionally relies on non-molecular data. For very fast-evolving organisms such as SARS-CoV-2, for which samples are obtained over a time span, the collection times provide this external information from which we can learn the rate of molecular evolution and date past events (Boni et al. 2020). In groups for which the fossil record is abundant, state-of-the-art dating methods use fossil information to complement molecular data, either in the form of a prior distribution on node ages (Nguyen & Ho 2020), or as data modelled with a fossilization process (Heath et al. 2014). Dating is a challenge in groups that lack fossils or other geological evidence, such as very old lineages and microbial lineages. In these groups, horizontal gene transfer (HGT) events have been identified as informative about relative dates: the ancestor of the gene's donor must be older than the descendants of the gene's recipient. Previous work using HGTs to date phylogenies have used methodologies that are ad-hoc (Davín et al 2018) or employ a small number of HGTs only (Magnabosco et al. 2018, Wolfe & Fournier 2018). Szöllősi et al. (2021) present and validate a Bayesian approach to estimate the age of diversification events based on relative information on these ages, such as implied by HGTs. This approach is flexible because it is modular: constraints on relative node ages can be combined with absolute age information from fossil data, and with any substitution model of molecular evolution, including complex state-of-art models. To ease the computational burden, the authors also introduce a two-step approach, in which the complexity of estimating branch lengths in substitutions per site is decoupled from the complexity of timing the tree with branch lengths in years, accounting for uncertainty in the first step. Currently, one limitation is that the tree topology needs to be known, and another limitation is that constraints need to be certain. Users of this method should be mindful of the latter when hundreds of constraints are used, as done by Szöllősi et al. (2021) to date the trees of Cyanobacteria and Archaea. Szöllősi et al. (2021)'s method is implemented in RevBayes, a highly modular platform for phylogenetic inference, rapidly growing in popularity (Höhna et al. 2016). The RevBayes tutorial page features a step-by-step tutorial "Dating with Relative Constraints", which makes the method highly approachable. References: Boni MF, Lemey P, Jiang X, Lam TT-Y, Perry BW, Castoe TA, Rambaut A, Robertson DL (2020) Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nature Microbiology, 5, 1408–1417. https://doi.org/10.1038/s41564-020-0771-4 Davín AA, Tannier E, Williams TA, Boussau B, Daubin V, Szöllősi GJ (2018) Gene transfers can date the tree of life. Nature Ecology & Evolution, 2, 904–909. https://doi.org/10.1038/s41559-018-0525-3 Heath TA, Huelsenbeck JP, Stadler T (2014) The fossilized birth–death process for coherent calibration of divergence-time estimates. Proceedings of the National Academy of Sciences, 111, E2957–E2966. https://doi.org/10.1073/pnas.1319091111 Höhna S, Landis MJ, Heath TA, Boussau B, Lartillot N, Moore BR, Huelsenbeck JP, Ronquist F (2016) RevBayes: Bayesian Phylogenetic Inference Using Graphical Models and an Interactive Model-Specification Language. Systematic Biology, 65, 726–736. https://doi.org/10.1093/sysbio/syw021 Magnabosco C, Moore KR, Wolfe JM, Fournier GP (2018) Dating phototrophic microbial lineages with reticulate gene histories. Geobiology, 16, 179–189. https://doi.org/10.1111/gbi.12273 Nguyen JMT, Ho SYW (2020) Calibrations from the Fossil Record. In: The Molecular Evolutionary Clock: Theory and Practice (ed Ho SYW), pp. 117–133. Springer International Publishing, Cham. https://doi.org/10.1007/978-3-030-60181-2_8 Szollosi, G.J., Hoehna, S., Williams, T.A., Schrempf, D., Daubin, V., Boussau, B. (2021) Relative time constraints improve molecular dating. bioRxiv, 2020.10.17.343889, ver. 8 recommended and peer-reviewed by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.10.17.343889 Wolfe JM, Fournier GP (2018) Horizontal gene transfer constrains the timing of methanogen evolution. Nature Ecology & Evolution, 2, 897–903. https://doi.org/10.1038/s41559-018-0513-7 | Relative time constraints improve molecular dating | Gergely J Szollosi, Sebastian Hoehna, Tom A Williams, Dominik Schrempf, Vincent Daubin, Bastien Boussau | <p style="text-align: justify;">Dating the tree of life is central to understanding the evolution of life on Earth. Molecular clocks calibrated with fossils represent the state of the art for inferring the ages of major groups. Yet, other informat... | | Bioinformatics & Computational Biology, Genome Evolution, Phylogenetics / Phylogenomics | Cécile Ané | 2020-10-21 23:39:17 | ||
08 Aug 2018

Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutationsInbreeding compensates for reduced sexual selection in purging deleterious mutationsRecommended by Charles Baer based on reviews by 2 anonymous reviewersTwo evolutionary processes have been shown in theory to enhance the effects of natural selection in purging deleterious mutations from a population (here ""natural"" selection is defined as ""selection other than sexual selection""). First, inbreeding, especially self-fertilization, facilitates the removal of deleterious recessive alleles, the effects of which are largely hidden from selection in heterozygotes when mating is random. Second, sexual selection can facilitate the removal of deleterious alleles of arbitrary dominance, with little or no demographic cost, provided that deleterious effects are greater in males than in females (""genic capture""). Inbreeding (especially selfing) and sexual selection are often negatively correlated in nature. Empirical tests of the role of sexual selection in purging deleterious mutations have been inconsistent, potentially due to the positive relationship between sexual selection and intersexual genetic conflict. References [1] Noël, E., Fruitet, E., Lelaurin, D., Bonel, N., Segard, A., Sarda, V., Jarne, P., & David P. (2018). Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations. bioRxiv, 273367, ver. 3 recommended and peer-reviewed by PCI Evol Biol. doi: 10.1101/273367 | Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations | E. Noël, E. Fruitet, D. Lelaurin, N. Bonel, A. Ségard, V. Sarda, P. Jarne and P. David | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (https://dx.doi.org/10.24072/pci.evolbiol.100055). Theory and empirical data showed that two processes can boost selection against deleterious mutations, ... | | Adaptation, Experimental Evolution, Reproduction and Sex, Sexual Selection | Charles Baer | Anonymous | 2018-03-01 08:12:37 | |
06 Sep 2022

Masculinization of the X-chromosome in aphid soma and gonadsSex-biased gene expression is not tissue-specific in Pea AphidsRecommended by Charles Baer and Tanja Schwander based on reviews by Ann Kathrin Huylmans and 1 anonymous reviewerSexual antagonism (SA), wherein the fitness interests of the sexes do not align, is inherent to organisms with two (or more) sexes. SA leads to intra-locus sexual conflict, where an allele that confers higher fitness in one sex reduces fitness in the other [1, 2]. This situation leads to what has been referred to as "gender load", resulting from the segregation of SA alleles in the population. Gender load can be reduced by the evolution of sex-specific (or sex-biased) gene expression. A specific prediction is that gene-duplication can lead to sub- or neo-functionalization, in which case the two duplicates partition the function in the different sexes. The conditions for invasion by a SA allele differ between sex-chromosomes and autosomes, leading to the prediction that (in XY or XO systems) the X should accumulate recessive male-favored alleles and dominant female-favored alleles; similar considerations apply in ZW systems ([3, but see 4]. Aphids present an interesting special case, for several reasons: they have XO sex-determination, and three distinct reproductive morphs (sexual females, parthenogenetic females, and males). Previous theoretical work by the lead author predict that the X should be optimized for male function, which was borne out by whole-animal transcriptome analysis [5]. Here [6], the authors extend that work to investigate “tissue”-specific (heads, legs and gonads), sex-specific gene expression. They argue that, if intra-locus SA is the primary driver of sex-biased gene expression, it should be generally true in all tissues. They set up as an alternative the possibility that sex-biased gene expression could also be driven by dosage compensation. They cite references supporting their argument that "dosage compensation (could be) stronger in the brain", although the underlying motivation for that argument appears to be based on empirical evidence rather than theoretical predictions. At any rate, the results are clear: all tissues investigated show masculinization of the X. Further, X-linked copies of gene duplicates were more frequently male-biased than duplicated autosomal genes or X-linked single-copy genes. To sum up, this is a nice empirical study with clearly interpretable (and interpreted) results, the most obvious of which is the greater sex-biased expression in sexually-dimorphic tissues. Unfortunately, as the authors emphasize, there is no general theory by which SA, variable dosage-compensation, and meiotic sex chromosome inactivation can be integrated in a predictive framework. It is to be hoped that empirical studies such as this one will motivate deeper and more general theoretical investigations. References [1] Rice WR, Chippindale AK (2001) Intersexual ontogenetic conflict. Journal of Evolutionary Biology 14: 685-693. https://doi.org/10.1046/j.1420-9101.2001.00319.x [2] Bonduriansky R, Chenoweth SF (2009) Intralocus sexual conflict. Trends Ecol Evol 24: 280-288. https://doi.org/10.1016/j.tree.2008.12.005 [3] Rice WR. (1984) Sex chromosomes and the evolution of sexual dimorphism. Evolution 38: 735-742. https://doi.org/10.1086/595754 [4] Fry JD (2010) The genomic location of sexually antagonistic variation: some cautionary comments. Evolution 64: 1510-1516. https://doi.org/10.1111%2Fj.1558-5646.2009.00898.x [5] Jaquiéry J, Rispe C, Roze D, Legeai F, Le Trionnaire G, Stoeckel S, et al. (2013) Masculinization of the X Chromosome in the Pea Aphid. PLoS Genetics 9. https://doi.org/10.1371/journal.pgen.1003690 [6] Jaquiéry J, Simon J-C, Robin S, Richard G, Peccoud J, Boulain H, Legeai F, Tanguy S, Prunier-Leterme N, Le Trionnaire G (2022) Masculinization of the X-chromosome in aphid soma and gonads. bioRxiv, 2021.08.13.453080, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.08.13.453080 | Masculinization of the X-chromosome in aphid soma and gonads | Julie Jaquiery, Jean-Christophe Simon, Stephanie Robin, Gautier Richard, Jean Peccoud, Helene Boulain, Fabrice Legeai, Sylvie Tanguy, Nathalie Prunier-Leterme, Gael Letrionnaire | <p>Males and females share essentially the same genome but differ in their optimal values for many phenotypic traits, which can result in intra-locus conflict between the sexes. Aphids display XX/X0 sex chromosomes and combine unusual X chromosome... | | Genetic conflicts, Genome Evolution, Reproduction and Sex | Charles Baer | 2021-08-16 08:56:08 | ||
16 May 2023
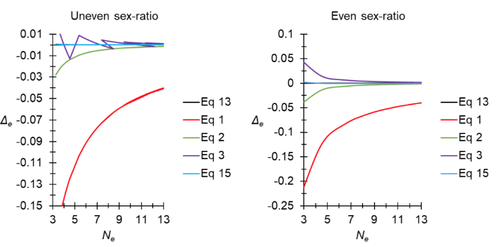
A new and almost perfectly accurate approximation of the eigenvalue effective population size of a dioecious population: comparisons with other estimates and detailed proofsAll you ever wanted to know about Ne in one handy placeRecommended by Charles Baer based on reviews by Jesse ("Jay") Taylor and 1 anonymous reviewerOf the four evolutionary forces, three can be straightforwardly summarized both conceptually and mathematically in the context of an allele at a genomic locus. Mutation (the mutation rate, μ) is simply captured by the per-site, per-generation probability that an allele mutates into a different allele. Recombination (the recombination rate, r) is captured as the probability of recombination between two sites, wherein alleles that are in different genomes in one generation come together in the same genome in the next generation. Natural selection (the selection coefficient, s) is captured by the probability that an allele is present in the next generation, relative to some reference. Random genetic drift – the random fluctuation in allele frequency due to sampling in a finite population - is not so straightforwardly summarized. The first, and most common way of characterizing evolutionary dynamics in a finite population is the Wright-Fisher model, in which the only deviation from the assumptions of Hardy-Weinberg conditions is finite population size. Importantly, in a W-F population, mating between diploid individuals is random, which implies self-fertile monoecy, and generations are non-overlapping. In an ideal W-F population, the probability that a gene copy leaves i descendants in the next generation is the result of binomial sampling of uniting gametes (if the locus is biallelic). The – and the next word is meaningful – magnitude/strength/rate/power/amount of genetic drift is proportional to 1/2N, where N is the size of the population. All of the following are affected by genetic drift: (1) the probability that a neutral allele ultimately reaches fixation, (2) the rate of loss of genetic variation within a population, (3) the rate of increase of genetic variance among populations, (4) the amount of genetic variation segregating in a population, (5) the probability of fixation/loss of a weakly selected variant. Presumably no real population adheres to ideal W-F conditions, which leads to the notion of "effective population size", Ne (Wright 1931), loosely defined as "the size of an ideal W-F population that experiences an equivalent strength of genetic drift". Almost always, Ne<N, and any violation of W-F assumptions can affect Ne. Importantly, Ne can be defined in different ways, and the specific formulation of Ne can have different implications for evolution. Ne was initially defined in terms of the rate of decrease of heterozygosity (inbreeding effective size) and increase in variance among populations (variance effective size). Ewens (1979) defined the Eigenvalue effective size (equivalent to the "random extinction" effective size) and elaborated on the conditions under which the various formulations of Ne differ (Ewens 1982). Nordborg and Krone (2002) defined the effective size in terms of the coalescent, and they identified conditions in which genetic drift cannot be described in terms of a W-F model (Sjodin et al. 2005); also see Karasov et al. (2010); Neher and Shraiman (2011). Distinct from the issue of defining Ne is the issue of calculating Ne from data, which is the focus of this paper by De Meeus and Noûs (2023). Pudovkin et al. (1996) showed that the Eigenvalue effective size in a dioecious population can be formulated in terms of excess heterozygosity, which the current authors note is equivalent to formulating Ne in terms of Wright's FIS statistic. As emphasized by the title, the marquee contribution of this paper is to provide a better approximation of the Eigenvalue effective size in a dioecious population. Science marches onward, although the empirical utility of this advance is obviously limited, given the tremendous inherent sources of uncertainty in real-world estimates of Ne. Perhaps more valuable, however, is the extensive set of appendixes, in which detailed derivations are provided for the various formulations of effective size. By way of analogy, the material presented here can be thought of as an extension of the material presented in section 7.6 of Crow and Kimura (1970), in which the Inbreeding and Variance effective population sizes are derived and compared. The appendixes should serve as a handy go-to source of detailed theoretical information with respect to the different formulations of effective population size. REFERENCES Crow, J. F. and M. Kimura. 1970. An Introduction to Population Genetics Theory. The Blackburn Press, Caldwell, NJ. De Meeûs, T. and Noûs, C. 2023. A new and almost perfectly accurate approximation of the eigenvalue effective population size of a dioecious population: comparisons with other estimates and detailed proofs. Zenodo, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.5281/zenodo.7927968 Ewens, W. J. 1979. Mathematical Population Genetics. Springer-Verlag, Berlin. Ewens, W. J. 1982. On the concept of the effective population size. Theoretical Population Biology 21:373-378. https://doi.org/10.1016/0040-5809(82)90024-7 Karasov, T., P. W. Messer, and D. A. Petrov. 2010. Evidence that adaptation in Drosophila Is not limited by mutation at single sites. Plos Genetics 6. https://doi.org/10.1371/journal.pgen.1000924 Neher, R. A. and B. I. Shraiman. 2011. Genetic Draft and Quasi-Neutrality in Large Facultatively Sexual Populations. Genetics 188:975-U370. https://doi.org/10.1534/genetics.111.128876 Nordborg, M. and S. M. Krone. 2002. Separation of time scales and convergence to the coalescent in structured populations. Pp. 194–232 in M. Slatkin, and M. Veuille, eds. Modern Developments in Theoretical Population Genetics: The Legacy of Gustave Malécot. Oxford University Press, Oxford. https://www.webpages.uidaho.edu/~krone/malecot.pdf Pudovkin, A. I., D. V. Zaykin, and D. Hedgecock. 1996. On the potential for estimating the effective number of breeders from heterozygote-excess in progeny. Genetics 144:383-387. https://doi.org/10.1093/genetics/144.1.383 Sjodin, P., I. Kaj, S. Krone, M. Lascoux, and M. Nordborg. 2005. On the meaning and existence of an effective population size. Genetics 169:1061-1070. https://doi.org/10.1534/genetics.104.026799 Wright, S. 1931. Evolution in Mendelian populations. Genetics 16:0097-0159. https://doi.org/10.1093/genetics/16.2.97 | A new and almost perfectly accurate approximation of the eigenvalue effective population size of a dioecious population: comparisons with other estimates and detailed proofs | Thierry de Meeûs and Camille Noûs | <p>The effective population size is an important concept in population genetics. It corresponds to a measure of the speed at which genetic drift affects a given population. Moreover, this is most of the time the only kind of population size that e... | | Bioinformatics & Computational Biology, Evolutionary Ecology, Evolutionary Theory, Population Genetics / Genomics, Reproduction and Sex | Charles Baer | 2023-02-22 16:53:49 | ||
28 Sep 2020

Evolution and genetic architecture of disassortative mating at a locus under heterozygote advantageEvolutionary insights into disassortative mating and its association to an ecologically relevant supergeneRecommended by Charles Mullon based on reviews by Tom Van Dooren and 2 anonymous reviewers based on reviews by Tom Van Dooren and 2 anonymous reviewers
Heliconius butterflies are famous for their colorful wing patterns acting as a warning of their chemical defenses [1]. Most species are involved in Müllerian mimicry assemblies, as predators learn to associate common wing patterns with unpalatability and preferentially target rare variants. Such positive-frequency dependent selection homogenizes wing patterns at different localities, and in several species, all individuals within a community belong to the same morph [2]. In this respect, H. numata stands out. This species shows stable local polymorphism across multiple localities, with local populations home to up to seven distinct morphs [2]. Although a balance between migration and local positive-frequency dependent selection can allow some degree of local polymorphism, theory suggests that this occurs only when migration is within a narrow window [3]. References [1] Merrill, R M, K K Dasmahapatra, J W Davey, D D Dell'Aglio, J J Hanly, B Huber, C D Jiggins, et al. (2015). The Diversification of Heliconius butterflies: What Have We Learned in 150 Years? Journal of Evolutionary Biology 28 (8), 1417–38. https://doi.org/10.1111/jeb.12672. | Evolution and genetic architecture of disassortative mating at a locus under heterozygote advantage | Ludovic Maisonneuve, Mathieu Joron, Mathieu Chouteau and Violaine Llaurens | <p>The evolution of mate preferences may depend on natural selection acting on the mating cues and on the underlying genetic architecture. While the evolution of assortative mating with respect to locally adapted traits has been well-characterized... | | Evolutionary Theory, Population Genetics / Genomics, Reproduction and Sex, Sexual Selection | Charles Mullon | 2019-10-29 09:55:18 | ||
23 Jan 2023
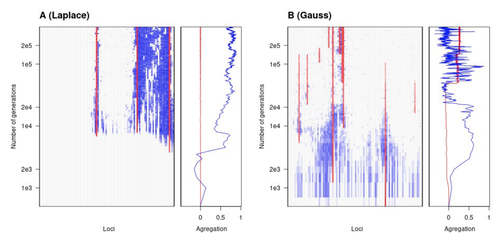
The genetic architecture of local adaptation in a clineEnvironmental and fitness landscapes matter for the genetic basis of local adaptationRecommended by Charles Mullon based on reviews by 2 anonymous reviewers
Natural landscapes are often composite, with spatial variation in environmental factors being the norm rather than exception. Adaptation to such variation is a major driver of diversity at all levels of biological organization, from genes to phenotypes, species and ultimately ecosystems. While natural selection favours traits that show a better fit to local conditions, the genomic response to such selection is not necessarily straightforward. This is because many quantitative traits are complex and the product of many loci, each with a small to moderate phenotypic contribution. Adapting to environmental challenges that occur in narrow ranges may thus prove difficult as each individual locus is easily swamped by alleles favoured across the rest of the population range. To better understand whether and how evolution overcomes such a hurdle, Laroche and Lenormand [1] combine quantitative genetics and population genetic modelling to track genomic changes that underpin a trait whose fitness optimum differs between a certain spatial range, referred to as a “pocket”, and the rest of the habitat. As it turns out from their analysis, one critical and probably underappreciated factor in determining the type of genetic architecture that evolves is how fitness declines away from phenotypic optima. One classical and popular model of fitness landscape that relates trait value to reproductive success is Gaussian, whereby small trait variations away from the optimum result in even smaller variations in fitness. This facilitates local adaptation via the invasion of alleles of small effects as carriers inside the pocket show a better fit while those outside the pocket only suffer a weak fitness cost. By contrast, when the fitness landscape is more peaked around the optimum, for instance where the decline is linear, adaptation through weak effect alleles is less likely, requiring larger pockets that are less easily swamped by alleles selected in the rest of the range. In addition to mathematically investigating the initial emergence of local adaptation, Laroche and Lenormand use computer simulations to look at its long-term maintenance. In principle, selection should favour a genetic architecture that consolidates the phenotype and increases its heritability, for instance by grouping several alleles of large effects close to one another on a chromosome to avoid being broken down by meiotic recombination. Whether or not this occurs also depends on the fitness landscape. When the landscape is Gaussian, the genetic architecture of the trait eventually consists of tightly linked alleles of large effects. The replacement of small effects by large effects loci is here again promoted by the slow fitness decline around the optimum. This is because any shift in architecture in an adapted population requires initially crossing a fitness valley. With a Gaussian landscape, this valley is shallow enough to be crossed, facilitated by a bit of genetic drift. By contrast, when fitness declines linearly around the optimum, genetic architecture is much less evolutionarily labile as any architecture change initially entails a fitness cost that is too high to bear. Overall, Laroche and Lenormand provide a careful and thought-provoking analysis of a classical problem in population genetics. In addition to questioning some longstanding modelling assumptions, their results may help understand why differentiated populations are sometimes characterized by “genomic islands” of divergence, and sometimes not. References [1] Laroche F, Lenormand T (2022) The genetic architecture of local adaptation in a cline. bioRxiv, 2022.06.30.498280, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.06.30.498280 | The genetic architecture of local adaptation in a cline | Fabien Laroche, Thomas Lenormand | <p>Local adaptation is pervasive. It occurs whenever selection favors different phenotypes in different environments, provided that there is genetic variation for the corresponding traits and that the effect of selection is greater than the effect... | | Adaptation, Evolutionary Theory, Genome Evolution, Molecular Evolution, Population Genetics / Genomics, Quantitative Genetics | Charles Mullon | 2022-07-07 08:46:47 |




