A behavior-manipulating virus relative as a source of adaptive genes for parasitoid wasps
D. Di Giovanni, D. Lepetit, M. Boulesteix, M. Ravallec, J. Varaldi
https://doi.org/10.1101/342758
Genetic intimacy of filamentous viruses and endoparasitoid wasps
Recommended by Ignacio Bravo based on reviews by Alejandro Manzano Marín and 1 anonymous reviewer
Viruses establish intimate relationships with the cells they infect. The virocell is a novel entity, different from the original host cell and beyond the mere combination of viral and cellular genetic material. In these close encounters, viral and cellular genomes often hybridise, combine, recombine, merge and excise. Such chemical promiscuity leaves genomics scars that can be passed on to descent, in the form of deletions or duplications and, importantly, insertions and back and forth exchange of genetic material between viruses and their hosts.
In this preprint [1], Di Giovanni and coworkers report the identification of 13 genes present in the extant genomes of members of the Leptopilina wasp genus, bearing sound signatures of having been horizontally acquired from an ancestral virus. Importantly the authors identify Leptopilina boulardi filamentous virus (LbFV) as an extant relative of the ancestral virus that served as donor for the thirteen horizontally transferred genes. While pinpointing genes with a likely possible viral origin in eukaryotic genomes is only relatively rare, identifying an extant viral lineage related to the ancestral virus that continues to infect an extant relative of the ancestral host is remarkable. But the amazing evolutionary history of the Leptopilina hosts and these filamentous viruses goes beyond this shared genes. These wasps are endoparasitoids of Drosophila larvae, the female wasp laying the eggs inside the larvae and simultaneously injecting venom that hinders the immune response. The composition of the venoms is complex, varies between wasp species and also between individuals within a species, but a central component of all these venoms are spiked structures that vary in morphology, symmetry and size, often referred to as virus-like particles (VLPs).
In this preprint, the authors convincingly show that the expression pattern in the Leptopilina wasps of the thirteen genes identified to have been horizontally acquired from the LbFV ancestor coincides with that of the production of VLPs in the female wasp venom gland. Based on this spatio-temporal match, the authors propose that these VLPs have a viral origin. The data presented in this preprint will undoubtedly stimulate further research on the composition, function, origin, evolution and diversity of these VLP structures, which are highly debated (see for instance [2] and [3]).
References
[1] Di Giovanni, D., Lepetit, D., Boulesteix, M., Ravallec, M., & Varaldi, J. (2018). A behavior-manipulating virus relative as a source of adaptive genes for parasitoid wasps. bioRxiv, 342758, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/342758
[2] Poirié, M., Colinet, D., & Gatti, J. L. (2014). Insights into function and evolution of parasitoid wasp venoms. Current Opinion in Insect Science, 6, 52-60. doi: 10.1016/j.cois.2014.10.004
[3] Heavner, M. E., Ramroop, J., Gueguen, G., Ramrattan, G., Dolios, G., Scarpati, M., ... & Govind, S. (2017). Novel organelles with elements of bacterial and eukaryotic secretion systems weaponize parasites of Drosophila. Current Biology, 27(18), 2869-2877. doi: 10.1016/j.cub.2017.08.019
| A behavior-manipulating virus relative as a source of adaptive genes for parasitoid wasps | D. Di Giovanni, D. Lepetit, M. Boulesteix, M. Ravallec, J. Varaldi | <p>To circumvent host immune response, numerous hymenopteran endo-parasitoid species produce virus-like structures in their reproductive apparatus that are injected into the host together with the eggs. These viral-like structures are absolutely n... |  | Adaptation, Behavior & Social Evolution, Genetic conflicts, Genome Evolution | Ignacio Bravo | | 2018-07-18 15:59:14 | View |
Replication-independent mutations: a universal signature ?
Recommended by Nicolas Galtier based on reviews by Marc Robinson-Rechavi and Robert Lanfear
Mutations are the primary source of genetic variation, and there is an obvious interest in characterizing and understanding the processes by which they appear. One particularly important question is the relative abundance, and nature, of replication-dependent and replication-independent mutations - the former arise as cells replicate due to DNA polymerization errors, whereas the latter are unrelated to the cell cycle. A recent experimental study in fission yeast identified a signature of mutations in quiescent (=non-replicating) cells: the spectrum of such mutations is characterized by an enrichment in insertions and deletions (indels) compared to point mutations, and an enrichment of deletions compared to insertions [2].
What Achaz et al. [1] report here is that the very same signature is detectable in humans. This time the approach is indirect and relies on two key aspects of mammalian reproduction biology: (1) oocytes remain quiescent over most of a female's lifespan, whereas spermatocytes keep dividing after male puberty, and (2) X chromosome, Y chromosome and autosomes spend different amounts of time in a female vs. male context. In agreement with the yeast study, Achaz et al. show that in humans the male-associated Y chromosome, for which quiescence is minimal, has by far the lowest ratios of indels to point mutations and of deletions to insertions, whereas the female-associated X chromosome has the highest. This is true both of variants that are polymorphic among humans and of fixed differences between humans and chimpanzees.
So we appear to be here learning about an important and general aspect of the mutation process. The authors suggest that, to a large extent, chromosomes tend to break in pieces at a rate that is proportional to absolute time - because indels in quiescent stage presumably result from double-strand DNA breaks. A very recent analysis of numerous mother-father-child trios in humans confirms this prediction in demonstrating an effect of maternal age, but not of paternal age, on the recombination rate [3]. This result also has important implications with respect to the interpretation of substitution rate variation among taxa and genomic compartments, particularly mitochondrial vs. nuclear, and their relationship with the generation time and longevity of organisms (e.g. [4]).
References
[1] Achaz, G., Gangloff, S., and Arcangioli, B. (2019). The quiescent X, the replicative Y and the Autosomes. BioRxiv, 351288, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/351288
[2] Gangloff, S., Achaz, G., Francesconi, S., Villain, A., Miled, S., Denis, C., and Arcangioli, B. (2017). Quiescence unveils a novel mutational force in fission yeast. eLife, 6:e27469. doi: 10.7554/eLife.27469
[3] Halldorsson, B. V., Palsson, G., Stefansson, O. A., Jonsson, H., Hardarson, M. T., Eggertsson, H. P., … Stefansson, K. (2019). Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science, 363: eaau1043. doi: 10.1126/science.aau1043
[4] Saclier, N., François, C. M., Konecny-Dupré, L., Lartillot, N., Guéguen, L., Duret, L., … Lefébure, T. (2019). Life History Traits Impact the Nuclear Rate of Substitution but Not the Mitochondrial Rate in Isopods. Molecular Biology and Evolution, in press. doi: 10.1093/molbev/msy247
| The quiescent X, the replicative Y and the Autosomes | Guillaume Achaz, Serge Gangloff, Benoit Arcangioli | <p>From the analysis of the mutation spectrum in the 2,504 sequenced human genomes from the 1000 genomes project (phase 3), we show that sexual chromosomes (X and Y) exhibit a different proportion of indel mutations than autosomes (A), ranking the... | 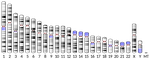 | Bioinformatics & Computational Biology, Genome Evolution, Human Evolution, Molecular Evolution, Population Genetics / Genomics, Reproduction and Sex | Nicolas Galtier | | 2018-07-25 10:37:48 | View |
Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisons
Sara Marin, Anaïs Gibert, Juliette Archambeau, Vincent Bonhomme, Mylène Lascoste and Benoit Pujol
https://doi.org/10.5281/zenodo.3628168
From populations to subspecies… to species? Contrasting patterns of local adaptation in closely-related taxa and their potential contribution to species divergence
Recommended by Emmanuelle Porcher based on reviews by Sophie Karrenberg, Santiago C. Gonzalez-Martinez and 1 anonymous reviewer
Elevation gradients are convenient and widely used natural setups to study local adaptation, particularly in these times of rapid climate change [e.g. 1]. Marin and her collaborators [2] did not follow the mainstream, however. Instead of tackling adaptation to climate change, they used elevation gradients to address another crucial evolutionary question [3]: could adaptation to altitude lead to ecological speciation, i.e. reproductive isolation between populations in spite of gene flow? More specifically, they examined how much local adaptation to environmental variation differed among closely-related, recently diverged subspecies. They studied several populations of two subspecies of snapdragon (Antirrhinum majus), with adjacent geographical distributions. Using common garden experiments and the classical, but still useful, QST-FST comparison, they demonstrate contrasting patterns of local adaptation to altitude between the two subspecies, with several traits under divergent selection in A. majus striatum but none in A. majus pseudomajus. These differences in local adaptation may contribute to species divergence, and open many stimulating questions on the underlying mechanisms, such as the identity of environmental drivers or contribution of reproductive isolation involving flower color polymorphism.
References
[1] Anderson, J. T., and Wadgymar, S. M. (2020). Climate change disrupts local adaptation and favours upslope migration. Ecology letters, 23(1), 181-192. doi: 10.1111/ele.13427
[2] Marin, S., Gibert, A., Archambeau, J., Bonhomme, V., Lascoste, M., and Pujol, B. (2020). Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisons. Zenodo, 3628168, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. doi: 10.5281/zenodo.3628168
[3] Schluter, D. (2009). Evidence for ecological speciation and its alternative. Science, 323(5915), 737-741. doi: 10.1126/science.1160006
| Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisons | Sara Marin, Anaïs Gibert, Juliette Archambeau, Vincent Bonhomme, Mylène Lascoste and Benoit Pujol | <p>Phenotypic divergence among natural populations can be explained by natural selection or by neutral processes such as drift. Many examples in the literature compare putatively neutral (FST) and quantitative genetic (QST) differentiation in mult... |  | Adaptation, Evolutionary Ecology, Genotype-Phenotype, Morphological Evolution, Quantitative Genetics | Emmanuelle Porcher | | 2018-08-05 15:34:30 | View |
Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pest
Georgios D. Koutsovoulos, Eder Marques, Marie-Jeanne Arguel, Laurent Duret, Andressa C.Z. Machado, Regina M.D.G. Carneiro, Djampa K. Kozlowski, Marc Bailly-Bechet, Philippe Castagnone-Sereno, Erika V.S. Albuquerque, Etienne G.J. Danchin
https://doi.org/10.1101/362129
The scandalous pest
Recommended by Nicolas Galtier based on reviews by 2 anonymous reviewers
Koutsovoulos et al. [1] have generated and analysed the first population genomic dataset in root-knot nematode Meloidogyne incognita. Why is this interesting? For two major reasons. First, M. incognita has been documented to be apomictic, i.e., to lack any form of sex. This is a trait of major evolutionary importance, with implications on species adaptive potential. The study of genome evolution in asexuals is fascinating and has the potential to inform on the forces governing the evolution of sex and recombination. Even small amounts of sex, however, are sufficient to restore most of the population genetic properties of true sexuals [2]. Because rare events of sex can remain undetected in the field, to confirm asexuality in M. incognita using genomic data is an important step. The second reason why M. incognita is of interest is that this nematode is one of the most harmful pests currently living on earth. M. incognita feeds on the roots of many cultivated plants, including tomato, bean, and cotton, and has been of major agricultural importance for decades. A number of races were defined based on host specificity. These have played a key role in attempts to control the dynamic of M. incognita populations via crop rotations. Races and management strategies so far lack any genetic basis, hence the second major interest of this study.
The authors newly sequenced the full genome of eleven strains from Brazil and added nine already available samples from Africa and North-America. They report that, in all likelihood, M. incognita is indeed a purely asexual species. This is supported by (i) the confirmation that the genome is in its major part haploid, and (ii) a spectacularly high level of linkage disequilibrium, which does not decline with genetic distance between loci at a 100kb scale. The absence of sex and recombination is associated in M. incognita with a remarkably low amount of genetic diversity - one order of magnitude less than in typical sexual nematodes - and an heavy load of deleterious mutations, as measured by the ratio of non-synonymous (=amino-acid changing) to synonymous (=amino-acid conservative) diversity in coding sequences. The other important result of this study is that the population substructure in M. incognita is in no way related to host races or geography. The tree genetic clusters that are identified include strains from several continents and feeding on a diversity of host plants.
The implications of this work are numerous. First, the results suggest that M. incognita is an ancient asexual. Asexuality, which was here demonstrated via linkage disequilibrium analysis, must be ancient enough for diploidy (or, in this case, maybe triploidy) to have been lost - i.e., formerly homologous chromosomes have accumulated enough mutations to be assembled as distinct entities. So we are not talking about a highly successful clone having recently spread the world - rather a long-term obligate parthenogen. Asexual organisms are deprived of the source of genetic variation offered by recombination, which is why asexuality is thought to be an evolutionary dead-end. Long-term asexuals are uncommon and even the most famous ones, bdelloid rotifers, are suspected to experience between-individual genetic transfers [3]. M. incognita is apparently a true 'evolutionary scandal', and as such deserves particular attention from molecular evolutionary geneticists.
The lack of any host race effect on the genetic diversity of M. incognita is another important finding. So-called 'races' have largely contributed to shape researchers' view of the structure of the species so far. This study demonstrates that a mental effort is now needed to forget about races, and consider host-specificity for what it is - a phenotypic trait. This result implies that many host shifts must have independently occurred in the three M. incognita genetic lineages, suggesting an arms race between plants and nematodes, which in the absence of sex and recombination must be entirely mutation-driven on the nematode side. Genes functionally involved in the arms race might therefore be expected to have experienced convergent evolution, if distinct M. incognita lineages have adopted the same solutions to overcome plant defenses. The present study paves the way for such a genome scan. The authors rightly discuss that the strong adaptive potential of M. incognita, at least in terms of host shift, despite no sex and tiny amounts of genetic diversity, is a paradox that would deserve to be further investigated.
References
[1] Koutsovoulos, G. D., Marques, E., Arguel, M. J., Duret, L., Machado, A. C. Z., Carneiro, R. M. D. G., Kozlowski, D. K., Bailly-Bechet, M., Castagnone-Sereno, P., Albuquerque, E. V., & Danchin, E. G. J. (2019). Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pest. bioRxiv, 362129, ver. 5, peer-reviewed and recommended by Peer Community in Evolutionary Biology. doi: 10.1101/362129
[2] Hartfield, M. (2016). Evolutionary genetic consequences of facultative sex and outcrossing. Journal of evolutionary biology, 29(1), 5-22. doi: 10.1111/jeb.12770
[3] Debortoli, N., Li, X., Eyres, I., Fontaneto, D., Hespeels, B., Tang, C. Q., Flot, J. F. & Van Doninck, K. (2016). Genetic exchange among bdelloid rotifers is more likely due to horizontal gene transfer than to meiotic sex. Current Biology, 26(6), 723-732. doi: 10.1016/j.cub.2016.01.031
| Population genomics supports clonal reproduction and multiple gains and losses of parasitic abilities in the most devastating nematode plant pest | Georgios D. Koutsovoulos, Eder Marques, Marie-Jeanne Arguel, Laurent Duret, Andressa C.Z. Machado, Regina M.D.G. Carneiro, Djampa K. Kozlowski, Marc Bailly-Bechet, Philippe Castagnone-Sereno, Erika V.S. Albuquerque, Etienne G.J. Danchin | <p>The most devastating nematodes to worldwide agriculture are the root-knot nematodes with Meloidogyne incognita being the most widely distributed and damaging species. This parasitic and ecological success seem surprising given its supposed obli... |  | Adaptation, Bioinformatics & Computational Biology, Evolutionary Ecology, Genome Evolution, Genotype-Phenotype, Molecular Evolution, Phylogenetics / Phylogenomics, Population Genetics / Genomics, Reproduction and Sex | Nicolas Galtier | | 2018-08-24 09:02:33 | View |
Evolution of selfing & lifespan 2.0
Recommended by Thomas Bataillon based on reviews by 2 anonymous reviewers
Flowering plants display a staggering diversity of both mating systems and life histories, ranging from almost exclusively selfers to obligate outcrossers, very short-lived annual herbs to super long lived trees. One pervasive pattern that has attracted considerable attention is the tight correlation that is found between mating systems and lifespan [1]. Until recently, theoretical explanations for these patterns have relied on static models exploring the consequences of several non-mutually exclusive important process: levels of inbreeding depression and ability to successfully were center stage. This make sense: successful colonization after long‐distance dispersal is far more likely to happen for self‐compatible than for self‐incompatible individuals in a sexually reproducing species. Furthermore, inbreeding depression (essentially a genetically driven phenomenon) and reproductive insurance are expected to shape the evolution of both mating system and lifespan.
But modelling jointly several processes and how their interplay to shape the evolution of a trait is challenging enough so models for the evolution of mating system tend invariably – for mathematical convenience and tractability – to fix lifespan [2].
However, comparative analysis of between species variations that map traits transitions among sister species in phylogenetic trees reveals a pervasive pattern: frequent transitions from a state outcrossing perennial to selfing annuals. This beg the question: is one transition triggering the other and if so, what comes first or are these transitions happening together?
In this work, Lesaffre and Billiard use a very sophisticated machinery developed by Kirkpatrick et al. [3] and consider a general class of so-called modifiers models [4]. They study jointly the evolution of life span and mating system. They do so by using models where different life stages are tracked with life stage having some (fixed for once) amount of inbreeding depression. Their paper is technically demanding, mixing analytics and computer simulations, and along the way generates several important findings that are expected to stimulate further empirical and theoretical studies: (1) pure selfing versus pure outcrossing is the expected stable evolutionary outcomes (despite observation that mixed mating systems can be regularly met in nature), (2) increasing life-span drastically reduces the scope for the evolution of selfing, conversely (3) transition to selfing will also select for shorter life span as a way to mitigate the cumulative effects of inbreeding depression on adult life stages.
As usual there is room for future work, in particular the authors’ model assumes fixed inbreeding depression in the different life stages and this highlights the need for models that explore how inbreeding depression, a pivotal quantity in these models, can itself be molded by both mating system and lifespan. A third-generation of models should be “soon” on the way!
References
[1] Grossenbacher D, Briscoe Runquist R, Goldberg EE, and Brandvain Y. (2015) Geographic range size is predicted by plant mating system. Ecology Letters 18, 706–713. doi: 10.1111/ele.12449
[2] Morgan MT, Schoen DJ, and Bataillon T. (1997) The evolution of self-fertilization in perennials. The American Naturalist 150, 618–638. doi: 10.1086/286085
[3] Kirkpatrick M, Johnson T, and Barton N. (2002) General models of multilocus evolution. Genetics 161, 1727–1750.
[4] Lesaffre, T, and Billiard S. (2019) The joint evolution of lifespan and self-fertilisation. bioRxiv, 420877, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/420877
| The joint evolution of lifespan and self-fertilisation | Thomas Lesaffre, Sylvain Billiard | <p>In Angiosperms, there exists a strong association between mating system and lifespan. Most self-fertilising species are short-lived and most predominant or obligate outcrossers are long-lived. This association is generally explained by the infl... |  | Evolutionary Theory, Life History, Reproduction and Sex | Thomas Bataillon | | 2018-09-19 10:03:51 | View |
Strong habitat and weak genetic effects shape the lifetime reproductive success in a wild clownfish population
Océane C. Salles, Glenn R. Almany, Michael L. Berumen, Geoffrey P. Jones, Pablo Saenz-Agudelo, Maya Srinivasan, Simon Thorrold, Benoit Pujol, Serge Planes
https://doi.org/10.5281/zenodo.3476529
Habitat variation of wild clownfish population shapes selfrecruitment more than genetic effects
Recommended by Philip Munday based on reviews by Juan Diego Gaitan-Espitia and Loeske Kruuk
Estimating the genetic and environmental components of variation in reproductive success is crucial to understanding the adaptive potential of populations to environmental change. To date, the heritability of lifetime reproductive success (fitness) has been estimated in a handful of wild animal population, mostly in mammals and birds, but has never been estimated for a marine species. The primary reason that such estimates are lacking in marine species is that most marine organisms have a dispersive larval phase, making it extraordinarily difficult to track the fate of offspring from one generation to the next.
In this study, Salles et al. [1] use an unprecedented 10 year data set for a wild population of orange clownfish (Amphiprion percula) to estimate the environmental, maternal and additive genetic components of life time reproductive success for the self-recruiting portion of the local population. Previous studies show that over 50% of juvenile clownfish recruiting to the population of clownfish at Kimbe Island (Kimbe Bay, PNG) are natal to the population. In other words, >50% of the juveniles recruiting to the population at Kimbe Island are offspring of parents from Kimbe Island. The identity and location of every adult clownfish in the Kimbe Island population was tracked over 10 years. At the same time newly recruiting juveniles were collected at regular intervals (biennially) and their parentage assigned with high confidence by 22 polymorphic microsatellite loci. Salles et al. then used a pedigree comprising 1735 individuals from up to 5 generations of clownfish at Kimbe Island to assess the contribution of every breeding pair of clownfish to self-recruitment within the local population. Because clownfish are site attached and live in close association with a host sea anemone, it was also possible to examine the contribution of reef location and host anemones species (either Heteractis magnifica or Stichodactyla gigantea) to reproductive success within the local population.
The study found that breeders from the eastern side of Kimbe Island, and mostly inhabiting S. gigantea sea anemones, produced more juveniles that recruited to the local population than breeders from other location around the island, or inhabiting H. magnifica. In fact, host anemone species and geographic location explained about 97% of the variance in reproductive success within the local population (i.e. excluding successful recruitment to other populations). By contrast, maternal and additive genetic effects explained only 1.9% and 1.3% of the variance, respectively. In other words, reef location and the species of host anemone inhabited had an overwhelming influence on the long-term contribution of breeding pairs of clownfish to replenishment of the local population. This overwhelming effect of the local habitat on reproductive success means that the population is potentially susceptible to rapid environmental changes - for example if S. giganta sea anemones are disproportionately susceptible to global warming, or reef habitats on the eastern side of the island are more susceptible to disturbance. By contrast, the small component of additive genetic variance in local reproductive success translated into low heritability and evolvability of lifetime reproductive success within the local population, as predicted by theory [2] and observed in some terrestrial species. Consequently, fitness would evolve slowly to environmental change.
Establishing the components of variation in fitness in a wild population of marine fishes is an astonishing achievement, made possible by the unprecedented long-term individual-level monitoring of the entire population of clownfish at Kimbe Island. A next step in this research would be to include other clownfish populations that are demographically and genetically connected to the Kimbe Island population through larval dispersal. It would be intriguing to establish the environmental, maternal and additive genetic components of reproductive success in the dispersing part of the Kimbe Island population, to see if this potentially differs among breeders who contribute more or less to replenishment within the local population.
References
[1] Salles, O. C., Almany, G. R., Berumen, M.L., Jones, G. P., Saenz-Agudelo, P., Srinivasan, M., Thorrold, S. R., Pujol, B., Planes, S. (2019). Strong habitat and weak genetic effects shape the lifetime reproductive success in a wild clownfish population. Zenodo, 3476529, ver. 3 peer-reviewed and recommended by Peer Community In Evolutionary Biology. doi: 10.5281/zenodo.3476529
[2] Fisher, R.A. (1930). The genetical theory of natural selection. Clarendon Press, Oxford, U.K.
| Strong habitat and weak genetic effects shape the lifetime reproductive success in a wild clownfish population | Océane C. Salles, Glenn R. Almany, Michael L. Berumen, Geoffrey P. Jones, Pablo Saenz-Agudelo, Maya Srinivasan, Simon Thorrold, Benoit Pujol, Serge Planes | <p>Lifetime reproductive success (LRS), the number of offspring an individual contributes to the next generation, is of fundamental importance in ecology and evolutionary biology. LRS may be influenced by environmental, maternal and additive genet... |  | Adaptation, Evolutionary Ecology, Life History, Quantitative Genetics | Philip Munday | | 2018-10-01 09:00:53 | View |
When sinks become sources: adaptive colonization in asexuals
Florian Lavigne, Guillaume Martin, Yoann Anciaux, Julien Papaïx, Lionel Roques
https://doi.org/10.1101/433235
Fisher to the rescue
Recommended by François Blanquart and Florence Débarre based on reviews by 3 anonymous reviewers
The ability of a population to adapt to a new niche is an important phenomenon in evolutionary biology. The colonisation of a new volcanic island by plant species; the colonisation of a host treated by antibiotics by a-resistant strain; the Ebola virus transmitting from bats to humans and spreading epidemically in Western Africa, are all examples of a population invading a new niche, adapting and eventually establishing in this new environment.
Adaptation to a new niche can be studied using source-sink models. In the original environment —the “source”—, the population enjoys a positive growth-rate and is self-sustaining, while in the new environment —the “sink”— the population has a negative growth rate and is able to sustain only by the continuous influx of migrants from the source. Understanding the dynamics of adaptation to the sink environment is challenging from a theoretical standpoint, because it requires modelling the demography of the sink as well as the transient dynamics of adaptation. Moreover, local selection in the sink and immigration from the source create distributions of genotypes that complicate the use of many common mathematical approaches.
In their paper, Lavigne et al. [1], develop a new deterministic model of adaptation to a harsh sink environment in an asexual species. The fitness of an individual is maximal when a number of phenotypes are tuned to an optimal value, and declines monotonously as phenotypes are further away from this optimum. This model —called Fisher’s Geometric Model— generates a GxE interaction for fitness because the phenotypic optimum in the sink environment is distinct from that in the source environment [2]. The authors circumvent mathematical difficulties by developing an original approach based on tracking the deterministic dynamics of the cumulant generating function of the fitness distribution in the sink. They derive a number of important results on the dynamics of adaptation to the sink:
From the point where immigration from the source to the sink starts, four phases of adaptation are observed. After a short transient phase (phase 1), a migration-selection balance is reached in the sink (phase 2). After a while, thanks to the immigration of rare adapted migrants and mutation in the sink, a small fraction of the sink population exhibits a close-to-optimal phenotype. This small adapted fraction grows in frequency and mean fitness rapidly increases in the sink (phase 3). Finally, the population settles around the sink optimum (phase 4) and, hurray, the sink is now a source! Interestingly, in this model the evolutionary dynamics do not depend on the immigration rate. In other words, adaptation will proceed at the same rate regardless of how many immigrants invade the sink. This is because the impact of immigration on adaptation depends on the rate of immigration relative to the sink density. This ratio is actually independent of immigration in a model where the sink is initially empty, migration from the sink back to the source is negligible and without density-dependence in the sink. In this model, mutation is a double-edged sword. Adapted phenotypes emerge from new mutations, and under this effect alone a higher mutation rate would translate into a shorter time to establishment in the sink. However, mutations may also have deleterious effects by displacing the phenotype away from the optimum. This mutation load will be greater when individuals need to simultaneously tune a large number of phenotypes. As a consequence of these two effects of mutations, time to establishment is minimal for an intermediate mutation rate. This result emerges from Fisher’s Geometric Model, but may hold more generally for biologically plausible fitness landscapes where mutations generates both beneficial (allowing adaptation to the sink) and deleterious genotypes. Lastly, in Fisher’s Geometric Model, the time to establishment increases superlinearly with harshness of the sink when the sink is too harsh, and establishment may occur only after a very long time. In these harsh sinks, the adapted genotypes are very few and increase very slowly in frequency, making the second phase of adaptation much longer. Thus, and as a direct consequence of Fisher’s Geometric Model, adding a “stepping stone” intermediate environment would allow faster adaptation to the extreme environment.
In conclusion, this theoretical work presents a method based on Fisher’s Geometric Model and the use of cumulant generating functions to resolve some aspects of adaptation to a sink environment. It generates a number of theoretical predictions for the adaptive colonisation of a sink by an asexual species with some standing genetic variation. It will be a fascinating task to examine whether these predictions hold in experimental evolution systems: will we observe the four phases of the dynamics of mean fitness in the sink environment? Will the rate of adaptation indeed be independent of the immigration rate? Is there an optimal rate of mutation for adaptation to the sink? Such critical tests of the theory will greatly improve our understanding of adaptation to novel environments.
References
[1] Lavigne, F., Martin, G., Anciaux, Y., Papaïx, J., and Roques, L. (2019). When sinks become sources: adaptive colonization in asexuals. bioRxiv, 433235, ver. 5 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/433235
[2] Martin, G., and Lenormand, T. (2006). A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species. Evolution, 60, 893-907. doi: 10.1111/j.0014-3820.2006.tb01169.x
| When sinks become sources: adaptive colonization in asexuals | Florian Lavigne, Guillaume Martin, Yoann Anciaux, Julien Papaïx, Lionel Roques | <p>The successful establishment of a population into a new empty habitat outside of its initial niche is a phenomenon akin to evolutionary rescue in the presence of immigration. It underlies a wide range of processes, such as biological invasions ... |  | Adaptation, Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology | François Blanquart | | 2018-10-03 20:59:16 | View |
Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network
Maud Irène Tenaillon, Khawla Sedikki, Maeva Mollion, Martine Le Guilloux, Elodie Marchadier, Adrienne Ressayre, Christine Dillmann
https://doi.org/10.1101/461947
Early and late flowering gene expression patterns in maize
Recommended by Tanja Pyhäjärvi based on reviews by Laura Shannon and 2 anonymous reviewers
Artificial selection experiments are key experiments in evolutionary biology. The demonstration that application of selective pressure across multiple generations results in heritable phenotypic changes is a tangible and reproducible proof of the evolution by natural selection.
Artificial selection experiments are used to evaluate the joint effects of selection on multiple traits, their genetic covariances and differences in responses in different environments. Most studies on artificial selection experiments report and base their analyses on phenotypic changes [1]. More recently, changes in allele frequency and other patterns of molecular genetic diversity have been used to identify genomic locations where selection has had an effect. However, so far the changes in gene expression have not been in the focus of artificial selection experiment studies (see [2] for an example though).
In plants, one of the most famous artificial selection experiments is the Illinois Corn Experiment where maize (Zea mays) is selected for oil and protein content [3], but in addition, similar experiments have been conducted also for other traits in maize. In Saclay divergent selection experiment [4] two maize inbred lines (F252 and MBS847) have been selected for early and late flowering for 13 generations, resulting in two week difference in flowering time.
In ”Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network ” [5] Maud Tenaillon and her coworkers study the gene expression differences among these two independently selected maize populations. Their experiments cover two years in field conditions and they use samples of shoot apical meristem at three different developmental stages: vegetative, transitioning and reproductive. They use RNA-seq transcriptome level differences and qRT-PCR for gene expression pattern investigation. The work is continuation to earlier genetic and phenotypic studies on the same material [4, 6].
The reviewers and I agree that dataset is unique and its major benefit is that it has been obtained from field conditions similar to those that species may face under natural setting during selection. Their tissue sampling is supported by flowering time phenotypic observations and covers the developmental transition stage, making a good effort to identify key transcriptional and phenotypic changes and their timing affected by selection.
Tenaillon et al. [5] identify more than 2000 genes that are differentially expressed among early and late flowering populations. Expectedly, they are enriched for known flowering time genes. As they point out, differential expression of thousands of genes does not mean that they all were independently affected by selection, but rather that the whole transcriptional network has shifted, possibly due to just few upstream or hub-genes. Also, the year-to-year variation had smaller effect in gene expression compared to developmental stage or genetic background, possibly indicating selection for stability across environmental fluctuation for such an important phenotype as flowering time.
Another noteworthy observation is that they find convergent patterns of transcriptional changes among the two selected lines. 115 genes expression patterns are shifted due to selection in both genetic backgrounds. This convergent pattern can be a result of either selection on standing variation or de novo mutations. The data does not allow testing which process is underlying the observed convergence. However, their results show that this is an interesting future question that can be addressed using genotype and gene expression data from the same ancestral and derived material and possibly their hybrids.
References
[1] Hill, W. G., & Caballero, A. (1992). Artificial selection experiments. Annual Review of Ecology and Systematics, 23(1), 287-310. doi: 10.1146/annurev.es.23.110192.001443
[2] Konczal, M., Babik, W., Radwan, J., Sadowska, E. T., & Koteja, P. (2015). Initial molecular-level response to artificial selection for increased aerobic metabolism occurs primarily through changes in gene expression. Molecular biology and evolution, 32(6), 1461-1473. doi: 10.1093/molbev/msv038
[3] Moose, S. P., Dudley, J. W., & Rocheford, T. R. (2004). Maize selection passes the century mark: a unique resource for 21st century genomics. Trends in plant science, 9(7), 358-364. doi: 10.1016/j.tplants.2004.05.005
[4] Durand, E., Tenaillon, M. I., Ridel, C., Coubriche, D., Jamin, P., Jouanne, S., Ressayre, A., Charcosset, A. and Dillmann, C. (2010). Standing variation and new mutations both contribute to a fast response to selection for flowering time in maize inbreds. BMC evolutionary biology, 10(1), 2. doi: 10.1186/1471-2148-10-2
[5] Tenaillon, M. I., Seddiki, K., Mollion, M., Le Guilloux, M., Marchadier, E., Ressayre, A. and Dillmann C. (2019). Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network. BioRxiv, 461947 ver. 5 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/461947
[6] Durand, E., Tenaillon, M. I., Raffoux, X., Thépot, S., Falque, M., Jamin, P., Bourgais A., Ressayre, A. and Dillmann, C. (2015). Dearth of polymorphism associated with a sustained response to selection for flowering time in maize. BMC evolutionary biology, 15(1), 103. doi: 10.1186/s12862-015-0382-5
| Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network | Maud Irène Tenaillon, Khawla Sedikki, Maeva Mollion, Martine Le Guilloux, Elodie Marchadier, Adrienne Ressayre, Christine Dillmann | <p>Artificial selection experiments are designed to investigate phenotypic evolution of complex traits and its genetic basis. Here we focused on flowering time, a trait of key importance for plant adaptation and life-cycle shifts. We undertook div... |  | Adaptation, Experimental Evolution, Expression Studies, Quantitative Genetics | Tanja Pyhäjärvi | | 2018-11-23 11:57:35 | View |
A bird’s white-eye view on neosex chromosome evolution
Thibault Leroy, Yoann Anselmetti, Marie-Ka Tilak, Sèverine Bérard, Laura Csukonyi, Maëva Gabrielli, Céline Scornavacca, Borja Milá, Christophe Thébaud, Benoit Nabholz
https://doi.org/10.1101/505610
Young sex chromosomes discovered in white-eye birds
Recommended by Kateryna Makova based on reviews by Gabriel Marais, Melissa Wilson and 1 anonymous reviewer
Recent advances in next-generation sequencing are allowing us to uncover the evolution of sex chromosomes in non-model organisms. This study [1] represents an example of this application to birds of two Sylvioidea species from the genus Zosterops (commonly known as white-eyes). The study is exemplary in the amount and types of data generated and in the thoroughness of the analysis applied. Both male and female genomes were sequenced to allow the authors to identify sex-chromosome specific scaffolds. These data were augmented by generating the transcriptome (RNA-seq) data set. The findings after the analysis of these extensive data are intriguing: neoZ and neoW chromosome scaffolds and their breakpoints were identified. Novel sex chromosome formation appears to be accompanied by translocation events. The timing of formation of novel sex chromosomes was identified using molecular dating and appears to be relatively recent. Yet first signatures of distinct evolutionary patterns of sex chromosomes vs. autosomes could be already identified. These include the accumulation of transposable elements and changes in GC content. The changes in GC content could be explained by biased gene conversion and altered recombination landscape of the neo sex chromosomes. The authors also study divergence and diversity of genes located on the neo sex chromosomes. Here their findings appear to be surprising and need further exploration. The neoW chromosome already shows unique patterns of divergence and diversity at protein-coding genes as compared with genes on either neoZ or autosomes. In contrast, the genes on the neoZ chromosome do not display divergence or diversity patterns different from those for autosomes. This last observation is puzzling and I believe should be explored in further studies. Overall, this study significantly advances our knowledge of the early stages of sex chromosome evolution in vertebrates, provides an example of how such a study could be conducted in other non-model organisms, and provides several avenues for future work.
References
[1] Leroy T., Anselmetti A., Tilak M.K., Bérard S., Csukonyi L., Gabrielli M., Scornavacca C., Milá B., Thébaud C. and Nabholz B. (2019). A bird’s white-eye view on neo-sex chromosome evolution. bioRxiv, 505610, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/505610
| A bird’s white-eye view on neosex chromosome evolution | Thibault Leroy, Yoann Anselmetti, Marie-Ka Tilak, Sèverine Bérard, Laura Csukonyi, Maëva Gabrielli, Céline Scornavacca, Borja Milá, Christophe Thébaud, Benoit Nabholz | <p>Chromosomal organization is relatively stable among avian species, especially with regards to sex chromosomes. Members of the large Sylvioidea clade however have a pair of neo-sex chromosomes which is unique to this clade and originate from a p... |  | Molecular Evolution, Population Genetics / Genomics | Kateryna Makova | | 2019-01-24 14:17:15 | View |
Fitting diversification models on undated or partially dated trees
Recommended by Nicolas Lartillot based on reviews by Amaury Lambert, Dominik Schrempf and 1 anonymous reviewer
Phylogenetic trees can be used to extract information about the process of diversification that has generated them. The most common approach to conduct this inference is to rely on a likelihood, defined here as the probability of generating a dated tree T given a diversification model (e.g. a birth-death model), and then use standard maximum likelihood. This idea has been explored extensively in the context of the so-called diversification studies, with many variants for the models and for the questions being asked (diversification rates shifting at certain time points or in the ancestors of particular subclades, trait-dependent diversification rates, etc).
However, all this assumes that the dated tree T is known without error. In practice, trees (that is, both the tree topology and the divergence times) are inferred based on DNA sequences, possibly combined with fossil information for calibrating and informing the divergence times. Molecular dating is a delicate exercise, however, and much more so in fact than reconstructing the tree topology. In particular, a mis-specificied model for the relaxed molecular clock, or a mis-specifiied prior, can have a substantial impact on the estimation of divergence dates - which in turn could severely mislead the inference about the underlying diversification process. This thus raises the following question: would that be possible to conduct inference and testing of diversification models without having to go through the dangerous step of molecular dating?
In his article ""Probabilities of tree topologies with temporal constraints and diversification shifts"" [1], Gilles Didier introduces a recursive method for computing the probability of a tree topology under some diversification model of interest, without knowledge of the exact dates, but only interval constraints on the dates of some of the nodes of the tree. Such interval constraints, which are derived from fossil knowledge, are typically used for molecular dating: they provide the calibrations for the relaxed clock analysis. Thus, what is essentially proposed by Gilles Didier is to use them in combination with the tree topology only, thus bypassing the need to estimates divergence times first, before fitting a diversification model to a phylogenetic tree.
This article, which is primarily a mathematical and algorithmic contribution, is then complemented with several applications: testing for a diversification shift in a given subclade of the phylogeny, just based on the (undated) tree topology, with interval constraints on some of its internal nodes; but also, computing the age distribution of each node and sampling on the joint distribution on node ages, conditional on the interval constraints. The test for the presence of a diversification shift is particularly interesting: an application to simulated data (and without any interval constraint in that case) suggests that the method based on the undated tree performs about as well as the classical method based on a dated tree, and this, even granting the classical approach a perfect knowledge of the dates - given that, in practice, one in fact relies on potentially biased estimates. Finally, an application to a well-known example (rate shifts in cetacean phylogeny) is presented.
This article thus represents a particularly meaningful contribution to the methodology for diversification studies; but also, for molecular dating itself: it is a well known problem in molecular dating that computing and sampling from the conditional distributions on node ages, given fossil constraints, and more generally understanding and visualizing how interval constraints on some nodes of the tree impact the distribution at other nodes, is a particularly difficult exercise. For that reason, the algorithmic routines presented in the present article will be useful in this context as well.
References
[1] Didier, G. (2020) Probabilities of tree topologies with temporal constraints and diversification shifts. bioRxiv, 376756, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/376756
| Probabilities of tree topologies with temporal constraints and diversification shifts | Gilles Didier | <p>Dating the tree of life is a task far more complicated than only determining the evolutionary relationships between species. It is therefore of interest to develop approaches apt to deal with undated phylogenetic trees. The main result of this ... |  | Bioinformatics & Computational Biology, Macroevolution | Nicolas Lartillot | | 2019-01-30 11:28:58 | View |







