
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields▲ | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
28 Feb 2018

Insects and incest: sib-mating tolerance in natural populations of a parasitoid waspIncestuous insects in nature despite occasional fitness costsRecommended by Caroline Nieberding and Bertanne Visser based on reviews by 2 anonymous reviewersInbreeding, or mating between relatives, generally lowers fitness [1]. Mating between genetically similar individuals can result in higher levels of homozygosity and consequently a higher frequency with which recessive disease alleles may be expressed within a population. Reduced fitness as a consequence of inbreeding, or inbreeding depression, can vary between individuals, sexes, populations and species [2], but remains a pervasive challenge for many organisms with small local population sizes, including humans [3]. But all is not lost for individuals within small populations, because an array of mechanisms can be employed to evade the negative effects of inbreeding [4], including sib-mating avoidance and dispersal [5, 6]. Despite thorough investigation of inbreeding and sib-mating avoidance in the laboratory, only very few studies have ventured into the field besides studies on vertebrates and eusocial insects. The study of Collet et al. [7] is a surprising exception, where the effect of male density and frequency of relatives on inbreeding avoidance was tested in the laboratory, after which robust field collections and microsatellite genotyping were used to infer relatedness and dispersal in natural populations. The parasitic wasp Venturia canescens is an excellent model system to study inbreeding, because mating success was previously found to decrease with increasing relatedness between mates in the laboratory [8] and this species thus suffers from inbreeding depression [9–11]. The authors used an elegant design combining population genetics and model simulations to estimate relatedness of mating partners in the field and compared that with a theoretical distribution of potential mate encounters when random mating is assumed. One of the most important findings of this study is that mating between siblings is not avoided in this species in the wild, despite negative fitness effects when inbreeding does occur. Similar findings were obtained for another insect species, the field cricket Gryllus campestris [12], which leaves us to wonder whether inbreeding tolerance could be more common in nature than currently appreciated. The authors further looked into sex-specific dispersal patterns between two patches located a few hundred meters apart. Females were indeed shown to be more related within a patch, but no genetic differences were observed between males, suggesting that V. canescens males more readily disperse. Moreover, microsatellite data at 18 different loci did not reveal genetic differentiation between populations approximately 300 kilometers apart. Gene flow is thus occurring over considerable distances, which could play an important role in the ability of this species to avoid negative fitness consequences of inbreeding in nature. Another interesting aspect of this work is that discrepancies were found between laboratory- and field-based data. What is the relevance of laboratory-based experiments if they cannot predict what is happening in the wild? Many, if not most, biologists (including us) bring our model system into the laboratory to control, at least to some extent, the plethora of environmental factors that could potentially affect our system (in ways that we do not want). Most behavioral studies on mating patterns and sexual selection are conducted in standardized laboratory conditions, but sexual selection is in essence social selection, because an individual’s fitness is partly determined by the phenotype of its social partners (i.e. the social environment) [13]. The social environment may actually dictate the expression of female mate choice and it is unclear how potential laboratory-induced social biases affect mating outcome. In V. canescens, findings using field-caught individuals paint a completely opposite picture of what was previously shown in the laboratory, i.e. sib-avoidance is not taking place in the field. It is likely that density, level of relatedness, sex ratio in the field, and/or the size of experimental arenas in the lab are all factors affecting mate selectivity, as we have previously shown in a butterfly [14–16]. If females, for example, typically only encounter a few males in sequence in the wild, it may be problematic for them to express choosiness when confronted simultaneously with two or more males in the laboratory. A recent study showed that, in the wild, female moths take advantage of staying in groups to blur male choosiness [17]. It is becoming more and more clear that what we observe in the laboratory may not actually reflect what is happening in nature [18]. Instead of ignoring the species-specific life history and ecological features of our favorite species when conducting lab experiments, we suggest that it is time to accept that we now have the theoretical foundations to tease apart what in this “environmental noise” actually shapes sexual selection in nature. Explicitly including ecology in studies on sexual selection will allow us to make more meaningful conclusions, i.e. rather than “this is what may happen in the wild”, we would be able to state “this is what often happens in nature”. References [1] Charlesworth D & Willis JH. 2009. The genetics of inbreeding depression. Nat. Rev. Genet. 10: 783–796. doi: 10.1038/nrg2664 | Insects and incest: sib-mating tolerance in natural populations of a parasitoid wasp | Marie Collet, Isabelle Amat, Sandrine Sauzet, Alexandra Auguste, Xavier Fauvergue, Laurence Mouton, Emmanuel Desouhant | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (http://dx.doi.org/10.24072/pci.evolbiol.100047) 1. Sib-mating avoidance is a pervasive behaviour that likely evolves in species subject to inbreeding dep... | | Behavior & Social Evolution, Evolutionary Ecology, Sexual Selection | Caroline Nieberding | 2017-07-28 09:23:20 | ||
12 Jul 2017

Despite reproductive interference, the net outcome of reproductive interactions among spider mite species is not necessarily costlyThe pros and cons of mating with strangersRecommended by Vincent Calcagno based on reviews by Joël Meunier and Michael D Greenfield
Interspecific matings are by definition rare events in nature, but when they occur they can be very important, and not only because they might condition gene flow between species. Even when such matings have no genetic consequence, for instance if they do not yield any fertile hybrid offspring, they can still have an impact on the population dynamics of the species involved [1]. Such atypical pairings between heterospecific partners are usually regarded as detrimental or undesired; as they interfere with the occurrence or success of intraspecific matings, they are expected to cause a decline in absolute fitness. References [1] Gröning J. & Hochkirch A. 2008. Reproductive interference between animal species. The Quarterly Review of Biology 83: 257-282. doi: 10.1086/590510 [2] Clemente SH, Santos I, Ponce AR, Rodrigues LR, Varela SAM & Magalhaes S. 2017 Despite reproductive interference, the net outcome of reproductive interactions among spider mite species is not necessarily costly. bioRxiv 113274, ver. 4 of the 30th of June 2017. doi: 10.1101/113274 | Despite reproductive interference, the net outcome of reproductive interactions among spider mite species is not necessarily costly | Salomé H. Clemente, Inês Santos, Rita Ponce, Leonor R. Rodrigues, Susana A. M. Varela and Sara Magalhães | Reproductive interference is considered a strong ecological force, potentially leading to species exclusion. This supposes that the net effect of reproductive interactions is strongly negative for one of the species involved. Testing this requires... | | Behavior & Social Evolution, Evolutionary Ecology, Species interactions | Vincent Calcagno | 2017-03-06 11:48:08 | ||
16 Dec 2016

POSTPRINT
Evolutionary robotics simulations help explain why reciprocity is rare in nature.Simulated robots and the evolution of reciprocityRecommended by Michael D Greenfield and Joël Meunier
Of the various forms of cooperative and altruistic behavior, reciprocity remains the most contentious. Humans certainly exhibit reciprocity – under certain circumstances – and various non-human animals behave in ways suggesting that they do as well. Thus, evolutionary biologists have sought to explain why non-relatives might engage in altruistic transactions when a substantial delay occurs between helping and compensation; i.e. an individual may be a donor today and a beneficiary tomorrow, or vice-versa. This quest, aided by game theory and computer modeling late in the past century, identified some strategies for reciprocal behavior that could work – in theory. But when biologists looked for confirmation of these strategies in animals they found little evidence that stood up to rigorous testing. In a recent paper André and Nolfi [1] offer a compelling reason for this observed rarity of reciprocity: Reciprocal behavior that animals might exhibit is a bit more complex than any of the game theoretic strategies, and even the simplest forms of realistic behavior would entail several nearly simultaneous mutations, an unlikely occurrence. André and Nolfi [1] relied on neural networks to test actors, robots that could evolve helping and reciprocal behavior from a basal level of selfishness. In an extensive series of simulations, they found that reciprocal behavior did not take hold in a population, largely because the various intermediates to full reciprocity were eliminated before the subsequent mutations occurred. The findings are satisfying given our current knowledge of animal behavior, but questions remain. Notably, how does one account for those rare cases in which reciprocity does meet all the criteria? The authors suggest some possibilities, but an analysis will await their next study. Reference [1] André J-B, Nolfi S. 2016. Evolutionary robotics simulations help explain why reciprocity is rare in nature. Scientific Reports 6:32785. doi: 10.1038/srep32785 | Evolutionary robotics simulations help explain why reciprocity is rare in nature. | André J-B, Nolfi S | The relative rarity of reciprocity in nature, contrary to theoretical predictions that it should be widespread, is currently one of the major puzzles in social evolution theory. Here we use evolutionary robotics to solve this puzzle. We show tha... | | Behavior & Social Evolution, Evolutionary Theory | Michael D Greenfield | 2016-12-16 18:08:31 | ||
13 Jan 2019
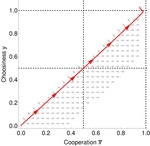
Why cooperation is not running awayA nice twist on partner choice theoryRecommended by Erol Akcay based on reviews by 2 anonymous reviewersIn this paper, Geoffroy et al. [1] deal with partner choice as a mechanism of maintaining cooperation, and argues that rather than being unequivocally a force towards improved payoffs to everyone through cooperation, partner choice can lead to “over-cooperation” where individuals can evolve to invest so much in cooperation that the costs of cooperating partially or fully negate the benefits from it. This happens when partner choice is consequential and effective, i.e., when interactions are long (so each decision to accept or reject a partner is a bigger stake) and when meeting new partners is frequent when unpaired (so that when one leaves an interaction one can find a new partner quickly). Geoffroy et al. [1] show that this tendency to select for overcooperation under such regimes can be counteracted if individuals base their acceptance-rejection of partners not just on the partner cooperativeness, but also on their own. By using tools from matching theory in economics, they show that plastic partner choice generates positive assortment between cooperativeness of the partners, and in the extreme case of perfectly assortative pairings, makes the pair the unit of selection, which selects for maximum total payoff. References [1] Geoffroy, F., Baumard, N., & Andre, J.-B. (2019). Why cooperation is not running away. bioRxiv, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/316117 | Why cooperation is not running away | Félix Geoffroy, Nicolas Baumard, Jean-Baptiste André | <p>A growing number of experimental and theoretical studies show the importance of partner choice as a mechanism to promote the evolution of cooperation, especially in humans. In this paper, we focus on the question of the precise quantitative lev... | | Behavior & Social Evolution, Evolutionary Theory | Erol Akcay | 2018-05-15 10:32:51 | ||
05 Feb 2021
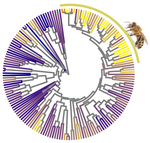
Relaxation of purifying selection suggests low effective population size in eusocial Hymenoptera and solitary pollinating beesMulti-gene and lineage comparative assessment of the strength of selection in HymenopteraRecommended by Bertanne Visser based on reviews by Michael Lattorff and 1 anonymous reviewerGenetic variation is the raw material for selection to act upon and the amount of genetic variation present within a population is a pivotal determinant of a population’s evolutionary potential. A large effective population size, i.e., the ideal number of individuals experiencing the same amount of genetic drift and inbreeding as an actual population, Ne (Wright 1931, Crow 1954), thus increases the probability of long-term survival of a population. However, natural populations, as opposed to theoretical ones, rarely adhere to the requirements of an ideal panmictic population (Sjödin et al. 2005). A range of circumstances can reduce Ne, including the structuring of populations (through space and time, as well as age and developmental stages) and inbreeding (Charlesworth 2009). In mammals, species with a larger body mass (as a proxy for lower Ne) were found to have a higher rate of nonsynonymous nucleotide substitutions (that alter the amino acid sequence of a protein), as well as radical amino acid substitutions (altering the physicochemical properties of a protein) (Popadin et al. 2007). In general, low effective population sizes increase the chance of mutation accumulation and drift, while reducing the strength of selection (Sjödin et al. 2005). References Charlesworth, B. (2009). Effective population size and patterns of molecular evolution and variation. Nature Reviews Genetics, 10(3), 195-205. doi: https://doi.org/10.1038/nrg2526 | Relaxation of purifying selection suggests low effective population size in eusocial Hymenoptera and solitary pollinating bees | Arthur Weyna, Jonathan Romiguier | <p>With one of the highest number of parasitic, eusocial and pollinator species among all insect orders, Hymenoptera features a great diversity of lifestyles. At the population genetic level, such life-history strategies are expected to decrease e... | | Behavior & Social Evolution, Genome Evolution, Life History, Molecular Evolution, Population Genetics / Genomics | Bertanne Visser | 2020-04-21 17:30:57 | ||
26 Nov 2019
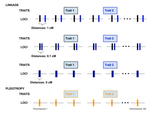
Pleiotropy or linkage? Their relative contributions to the genetic correlation of quantitative traits and detection by multi-trait GWA studiesUnderstanding the effects of linkage and pleiotropy on evolutionary adaptationRecommended by Kathleen Lotterhos based on reviews by Pär Ingvarsson and 1 anonymous reviewerGenetic correlations among traits are ubiquitous in nature. However, we still have a limited understanding of the genetic architecture of trait correlations. Some genetic correlations among traits arise because of pleiotropy - single mutations or genotypes that have effects on multiple traits. Other genetic correlations among traits arise because of linkage among mutations that have independent effects on different traits. Teasing apart the differential effects of pleiotropy and linkage on trait correlations is difficult, because they result in very similar genetic patterns. However, understanding these differential effects gives important insights into how ubiquitous pleiotropy may be in nature. References [1] Chebib, J. and Guillaume, F. (2019). Pleiotropy or linkage? Their relative contributions to the genetic correlation of quantitative traits and detection by multi-trait GWA studies. bioRxiv, 656413, v3 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/656413 | Pleiotropy or linkage? Their relative contributions to the genetic correlation of quantitative traits and detection by multi-trait GWA studies | Jobran Chebib and Frédéric Guillaume | <p>Genetic correlations between traits may cause correlated responses to selection depending on the source of those genetic dependencies. Previous models described the conditions under which genetic correlations were expected to be maintained. Sel... | | Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Dynamics, Evolutionary Theory, Genome Evolution, Genotype-Phenotype, Molecular Evolution, Population Genetics / Genomics, Quantitative Genetics | Kathleen Lotterhos | 2019-06-05 13:51:43 | ||
17 Dec 2016
POSTPRINT
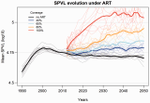
Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling studyPredicting HIV virulence evolution in response to widespread treatmentRecommended by Samuel Alizon and Roger Kouyos
It is a classical result in the virulence evolution literature that treatments decreasing parasite replication within the host should select for higher replication rates, thus driving increased levels of virulence if the two are correlated. There is some evidence for this in vitro but very little in the field. HIV infections in humans offer a unique opportunity to go beyond the simple predictions that treatments should favour more virulent strains because many details of this host-parasite system are known, especially the link between set-point virus load, transmission rate and virulence. To tackle this question, Herbeck et al. [1] used a detailed individual-based model. This is original because it allows them to integrate existing knowledge from the epidemiology and evolution of HIV (e.g. recent estimates of the ‘heritability’ of set-point virus load from one infection to the next). This detailed model allows them to formulate predictions regarding the effect of different treatment policies; especially regarding the current policy switch away from treatment initiation based on CD4 counts towards universal treatment. The results show that, perhaps as expected from the theory, treatments based on the level of remaining host target cells (CD4 T cells) do not affect virulence evolution because they do not strongly affect the virulence level that maximizes HIV’s transmission potential. However, early treatments can lead to moderate increase in virulence within several years if coverage is high enough. These results seem quite robust to variation of all the parameters in realistic ranges. The great step forward in this model is the ability to obtain quantitative prediction regarding how a virus may evolve in response to public health policies. Here the main conclusion is that given our current knowledge in HIV biology, the risk of virulence evolution is perhaps more limited than expected from a direct application of virulence evolution model. Interestingly, the authors also conclude that recently observed increased in HIV virulence [2-3] cannot be explained by the impact of antiretroviral therapy alone; which raises the question about the main mechanism behind this increase. Finally, the authors make the interesting suggestion that “changing virulence is amenable to being monitored alongside transmitted drug resistance in sentinel surveillance”. References [1] Herbeck JT, Mittler JE, Gottlieb GS, Goodreau SM, Murphy JT, Cori A, Pickles M, Fraser C. 2016. Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling study. Virus Evolution 2:vew028. doi: 10.1093/ve/vew028 [2] Herbeck JT, Müller V, Maust BS, Ledergerber B, Torti C, et al. 2012. Is the virulence of HIV changing? A meta-analysis of trends in prognostic markers of HIV disease progression and transmission. AIDS 26:193-205. doi: 10.1097/QAD.0b013e32834db418 [3] Pantazis N, Porter K, Costagliola D, De Luca A, Ghosn J, et al. 2014. Temporal trends in prognostic markers of HIV-1 virulence and transmissibility: an observational cohort study. Lancet HIV 1:e119-26. doi: 10.1016/s2352-3018(14)00002-2 | Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling study | Herbeck JT, Mittler JE, Gottlieb GS, Goodreau SM, Murphy JT, Cori A, Pickles M, Fraser C | There are global increases in the use of HIV antiretroviral therapy (ART), guided by clinical benefits of early ART initiation and the efficacy of treatment as prevention of transmission. Separately, it has been shown theoretically and empirically... | | Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Epidemiology | Samuel Alizon | 2016-12-16 20:54:08 | ||
21 Nov 2018
Convergent evolution as an indicator for selection during acute HIV-1 infectionIs convergence an evidence for positive selection?Recommended by Guillaume Achaz based on reviews by Jeffrey Townsend and 1 anonymous reviewerThe preprint by Bertels et al. [1] reports an interesting application of the well-accepted idea that positively selected traits (here variants) can appear several times independently; think about the textbook examples of flight capacity. Hence, the authors assume that reciprocally convergence implies positive selection. The methodology becomes then, in principle, straightforward as one can simply count variants in independent datasets to detect convergent mutations. References [1] Bertels, F., Metzner, K. J., & Regoes R. R. (2018). Convergent evolution as an indicator for selection during acute HIV-1 infection. BioRxiv, 168260, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/168260 | Convergent evolution as an indicator for selection during acute HIV-1 infection | Frederic Bertels, Karin J Metzner, Roland R Regoes | <p>Convergent evolution describes the process of different populations acquiring similar phenotypes or genotypes. Complex organisms with large genomes only rarely and only under very strong selection converge to the same genotype. In contrast, ind... | | Bioinformatics & Computational Biology, Evolutionary Applications, Genome Evolution, Molecular Evolution | Guillaume Achaz | 2017-07-26 08:39:17 | ||
17 Nov 2017

ABC random forests for Bayesian parameter inferenceMachine learning methods are useful for Approximate Bayesian Computation in evolution and ecologyRecommended by Michael Blum based on reviews by Dennis Prangle and Michael BlumIt is my pleasure to recommend the paper by Raynal et al. [1] about using random forest for parameter inference. There are two reviews about the paper, one review written by Dennis Prangle and another review written by myself. Both reviews were positive and included comments that have been addressed in the current version of the preprint. The paper nicely shows that modern machine learning approaches are useful for Approximate Bayesian Computation (ABC) and more generally for simulation-driven parameter inference in ecology and evolution. The authors propose to consider the random forest approach, proposed by Meinshausen [2] to perform quantile regression. The numerical implementation of ABC with random forest, available in the abcrf package, is based on the RANGER R package that provides a fast implementation of random forest for high-dimensional data. According to my reading of the manuscript, there are 3 main advantages when using random forest (RF) for parameter inference with ABC. The first advantage is that RF can handle many summary statistics and that dimension reduction is not needed when using RF. The second advantage is very nicely displayed in Figure 5, which shows the main result of the paper. If correct, 95% posterior credibility intervals (C.I.) should contain 95% of the parameter values used in simulations. Figure 5 shows that posterior C.I. obtained with rejection are too large compared to other methods. By contrast, C.I. obtained with regression methods have been shrunken. However, the shrinkage can be excessive for the smallest tolerance rates, with coverage values that can be equal to 85% instead of the expected 95% value. The attractive property of RF is that C.I. have been shrunken but the coverage is of 100% resulting in a conservative decision about parameter values. The last advantage is that no hyperparameter should be chosen. It is a parameter free approach, which is desirable because of the potential difficulty of choosing an appropriate acceptance rate. The main drawback of the proposed approach concerns joint parameter inference. There are many settings where the joint parameter distribution is of interest and the proposed RF approach cannot handle that. In population genetics for example, estimation of the severity and of the duration of the bottleneck should be estimated jointly because of identifiability issues. The challenge of performing joint parameter inference with RF might constitute a useful research perspective. References [1] Raynal L, Marin J-M, Pudlo P, Ribatet M, Robert CP, Estoup A. 2017. ABC random forests for Bayesian parameter inference. arXiv 1605.05537v4, https://arxiv.org/pdf/1605.05537 | ABC random forests for Bayesian parameter inference | Louis Raynal, Jean-Michel Marin, Pierre Pudlo, Mathieu Ribatet, Christian P. Robert, Arnaud Estoup | This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (http:// dx.doi.org/ 10.24072/ pci.evolbiol.100036). Approximate Bayesian computation (ABC) has grown into a standard methodology that manages Bayesian infer... | | Bioinformatics & Computational Biology, Evolutionary Applications, Other, Population Genetics / Genomics | Michael Blum | 2017-07-06 07:42:00 | ||
20 Jan 2020

A young age of subspecific divergence in the desert locust Schistocerca gregaria, inferred by ABC Random ForestEstimating recent divergence history: making the most of microsatellite data and Approximate Bayesian Computation approachesRecommended by Takeshi Kawakami and Concetta Burgarella based on reviews by Michael D Greenfield and 2 anonymous reviewersThe present-day distribution of extant species is the result of the interplay between their past population demography (e.g., expansion, contraction, isolation, and migration) and adaptation to the environment. Shedding light on the timing and magnitude of key demographic events helps identify potential drivers of such events and interaction of those drivers, such as life history traits and past episodes of environmental shifts. The understanding of the key factors driving species evolution gives important insights into how the species may respond to changing conditions, which can be particularly relevant for the management of harmful species, such as agricultural pests (e.g. [1]). Meaningful demographic inferences present major challenges. These include formulating evolutionary scenarios fitting species biology and the eco-geographical context and choosing informative molecular markers and accurate quantitative approaches to statistically compare multiple demographic scenarios and estimate the parameters of interest. A further issue comes with result interpretation. Accurately dating the inferred events is far from straightforward since reliable calibration points are necessary to translate the molecular estimates of the evolutionary time into absolute time units (i.e. years). This can be attempted in different ways, such as by using fossil and archaeological records, heterochronous samples (e.g. ancient DNA), and/or mutation rate estimated from independent data (e.g. [2], [3] for review). Nonetheless, most experimental systems rarely meet these conditions, hindering the comprehensive interpretation of results. The contribution of Chapuis et al. [4] addresses these issues to investigate the recent history of the African insect pest Schistocerca gregaria (desert locust). They apply Approximate Bayesian Computation-Random Forest (ABC-RF) approaches to microsatellite markers. Owing to their fast mutation rate microsatellite markers offer at least two advantages: i) suitability for analyzing recently diverged populations, and ii) direct estimate of the germline mutation rate in pedigree samples. The work of Chapuis et al. [4] benefits of both these advantages, since they have estimates of mutation rate and allele size constraints derived from germline mutations in the species [5]. The main aim of the study is to infer the history of divergence of the two subspecies of the desert locust, which have spatially disjoint distribution corresponding to the dry regions of North and West-South Africa. They first use paleo-vegetation maps to formulate hypotheses about changes in species range since the last glacial maximum. Based on them, they generate 12 divergence models. For the selection of the demographic model and parameter estimation, they apply the recently developed ABC-RF approach, a powerful inferential tool that allows optimizing the use of summary statistics information content, among other advantages [6]. Some methodological novelties are also introduced in this work, such as the computation of the error associated with the posterior parameter estimates under the best scenario. The accuracy of timing estimate is assured in two ways: i) by the use of microsatellite markers with known evolutionary dynamics, as underlined above, and ii) by assessing the divergence time threshold above which posterior estimates are likely to be biased by size homoplasy and limits in allele size range [7]. The best-supported model suggests a recent divergence event of the subspecies of S. gregaria (around 2.6 kya) and a reduction of populations size in one of the subspecies (S. g. flaviventris) that colonized the southern distribution area. As such, results did not support the hypothesis that the southward colonization was driven by the expansion of African dry environments associated with the last glacial maximum, as it has been postulated for other arid-adapted species with similar African disjoint distributions [8]. The estimated time of divergence points at a much more recent origin for the two subspecies, during the late Holocene, in a period corresponding to fairly stable arid conditions similar to current ones [9,10]. Although the authors cannot exclude that their microsatellite data bear limited information on older colonization events than the last one, they bring arguments in favour of alternative explanations. The hypothesis privileged does not involve climatic drivers, but the particularly efficient dispersal behaviour of the species, whose individuals are able to fly over long distances (up to thousands of kilometers) under favourable windy conditions. A single long-distance dispersal event by a few individuals would explain the genetic signature of the bottleneck. There is a growing number of studies in phylogeography in arid regions in the Southern hemisphere, but the impact of past climate changes on the species distribution in this region remains understudied relative to the Northern hemisphere [11,12]. The study presented by Chapuis et al. [4] offers several important insights into demographic changes and the evolutionary history of an agriculturally important pest species in Africa, which could also mirror the history of other organisms in the continent. As the authors point out, there are necessarily some uncertainties associated with the models of past ecosystems and climate, especially for Africa. Interestingly, the authors argue that the information on paleo-vegetation turnover was more informative than climatic niche modeling for the purpose of their study since it made them consider a wider range of bio-geographical changes and in turn a wider range of evolutionary scenarios (see discussion in Supplementary Material). Microsatellite markers have been offering a useful tool in population genetics and phylogeography for decades, but their popularity is perhaps being taken over by single nucleotide polymorphism (SNP) genotyping and whole-genome sequencing (WGS) (the peak year of the number of the publication with “microsatellite” is in 2012 according to PubMed). This study reaffirms the usefulness of these classic molecular markers to estimate past demographic events, especially when species- and locus-specific microsatellite mutation features are available and a powerful inferential approach is adopted. Nonetheless, there are still hurdles to overcome, such as the limitations in scenario choice associated with the simulation software used (e.g. not allowing for continuous gene flow in this particular case), which calls for further improvement of simulation tools allowing for more flexible modeling of demographic events and mutation patterns. In sum, this work not only contributes to our understanding of the makeup of the African biodiversity but also offers a useful statistical framework, which can be applied to a wide array of species and molecular markers (microsatellites, SNPs, and WGS). References [1] Lehmann, P. et al. (2018). Complex responses of global insect pests to climate change. bioRxiv, 425488. doi: https://dx.doi.org/10.1101/425488 [2] Donoghue, P. C., & Benton, M. J. (2007). Rocks and clocks: calibrating the Tree of Life using fossils and molecules. Trends in Ecology & Evolution, 22(8), 424-431. doi: https://dx.doi.org/10.1016/j.tree.2007.05.005 [3] Ho, S. Y., Lanfear, R., Bromham, L., Phillips, M. J., Soubrier, J., Rodrigo, A. G., & Cooper, A. (2011). Time‐dependent rates of molecular evolution. Molecular ecology, 20(15), 3087-3101. doi: https://dx.doi.org/10.1111/j.1365-294X.2011.05178.x [4] Chapuis, M.-P., Raynal, L., Plantamp, C., Meynard, C. N., Blondin, L., Marin, J.-M. and Estoup, A. (2020). A young age of subspecific divergence in the desert locust Schistocerca gregaria, inferred by ABC Random Forest. bioRxiv, 671867, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: https://dx.doi.org/10.1101/671867 5] Chapuis, M.-P., Plantamp, C., Streiff, R., Blondin, L., & Piou, C. (2015). Microsatellite evolutionary rate and pattern in Schistocerca gregaria inferred from direct observation of germline mutations. Molecular ecology, 24(24), 6107-6119. doi: https://dx.doi.org/10.1111/mec.13465 [6] Raynal, L., Marin, J. M., Pudlo, P., Ribatet, M., Robert, C. P., & Estoup, A. (2018). ABC random forests for Bayesian parameter inference. Bioinformatics, 35(10), 1720-1728. doi: https://dx.doi.org/10.1093/bioinformatics/bty867 [7] Estoup, A., Jarne, P., & Cornuet, J. M. (2002). Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Molecular ecology, 11(9), 1591-1604. doi: https://dx.doi.org/10.1046/j.1365-294X.2002.01576.x [8] Moodley, Y. et al. (2018). Contrasting evolutionary history, anthropogenic declines and genetic contact in the northern and southern white rhinoceros (Ceratotherium simum). Proceedings of the Royal Society B, 285(1890), 20181567. doi: https://dx.doi.org/10.1098/rspb.2018.1567 [9] Kröpelin, S. et al. (2008). Climate-driven ecosystem succession in the Sahara: the past 6000 years. science, 320(5877), 765-768. doi: https://dx.doi.org/10.1126/science.1154913 [10] Maley, J. et al. (2018). Late Holocene forest contraction and fragmentation in central Africa. Quaternary Research, 89(1), 43-59. doi: https://dx.doi.org/10.1017/qua.2017.97 [11] Beheregaray, L. B. (2008). Twenty years of phylogeography: the state of the field and the challenges for the Southern Hemisphere. Molecular Ecology, 17(17), 3754-3774. doi: https://dx.doi.org/10.1111/j.1365-294X.2008.03857.x [12] Dubey, S., & Shine, R. (2012). Are reptile and amphibian species younger in the Northern Hemisphere than in the Southern Hemisphere?. Journal of evolutionary biology, 25(1), 220-226. doi: https://dx.doi.org/10.1111/j.1420-9101.2011.02417.x ***** A video about this preprint is available here: | A young age of subspecific divergence in the desert locust Schistocerca gregaria, inferred by ABC Random Forest | Marie-Pierre Chapuis, Louis Raynal, Christophe Plantamp, Christine N. Meynard, Laurence Blondin, Jean-Michel Marin, Arnaud Estoup | <p>Dating population divergence within species from molecular data and relating such dating to climatic and biogeographic changes is not trivial. Yet it can help formulating evolutionary hypotheses regarding local adaptation and future responses t... | | Bioinformatics & Computational Biology, Evolutionary Applications, Phylogeography & Biogeography, Population Genetics / Genomics | Takeshi Kawakami | 2019-06-20 10:31:15 |




