
Latest recommendations

| Id | Title▼ | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
04 Jun 2019

Thermal regimes, but not mean temperatures, drive patterns of rapid climate adaptation at a continent-scale: evidence from the introduced European earwig across North AmericaTemperature variance, rather than mean, drives adaptation to local climateRecommended by Fabien Aubret based on reviews by Ben Phillips and Eric GangloffClimate change is impacting eco-systems worldwide and driving many populations to move, adapt or go extinct. It is increasingly appreciated, for example, that species may adjust their phenology in response to climate change, although empirical data is scarce. In this preprint [1], Tourneur and Meunier report an impressive sampling effort in which life-history traits were measured across introduced populations of earwig in North America. The authors examine whether variation in life-history across populations is correlated with aspects of the thermal climate experienced by each population: mean temperature and seasonality of temperature. They find some fascinating correlations between life-history and thermal climate; correlations with the seasonality of temperature, but not with mean temperature. This study provides relatively uncommon data, in the sense that where most of the literature looking at adaptation in animals in response to climate change has focused on physiological traits [2, 3], this study examines changes in life-history traits with time scales relevant to impending climate change, and provides a reasonable argument that this is adaptation, not just constraint. References [1] Tourneur, J.-C. and Meunier, J. (2019). Thermal regimes, but not mean temperatures, drive patterns of rapid climate adaptation at a continent-scale: evidence from the introduced European earwig across North America. BioRxiv, 550319, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/550319 | Thermal regimes, but not mean temperatures, drive patterns of rapid climate adaptation at a continent-scale: evidence from the introduced European earwig across North America | Jean-Claude Tourneur, Joël Meunier | <p>The recent development of human societies has led to major, rapid and often inexorable changes in the environment of most animal species. Over the last decades, a growing number of studies formulated predictions on the modalities of animal adap... | | Adaptation, Evolutionary Ecology, Life History | Fabien Aubret | 2019-02-15 09:12:11 | ||
14 Dec 2016

POSTPRINT
The Red Queen lives: epistasis between linked resistance lociEvidence of epistasis provides further support to the Red Queen theory of host-parasite coevolutionRecommended by Adele Mennerat and Thierry LefèvreAccording to the Red Queen theory of antagonistic host-parasite coevolution, adaptation of parasites to the most common host genotype results in negative frequency-dependent selection whereby rare host genotypes are favoured. Assuming that host resistance relies on a genetic host-parasite (mis)match involving several linked loci, then recombination appears as much more efficient than parthenogenesis in generating new resistant host genotypes. This has long been proposed to explain one of the biggest so-called paradoxes in evolutionary biology, i.e. the maintenance of recombination despite its twofold cost. Evidence from various study systems indicates that successful infection (and hence host resistance) depends on a genetic match between the parasite’s and the host’s genotype via molecular interactions involving elicitor/receptor mechanisms. However the key assumption of epistasis, i.e. that this genetic host-parasite match involves several linked resistance loci, remained unsupported so far. Metzger and coauthors [1] now provide empirical support for it. Daphnia magna can reproduce both sexually and clonally and their well-studied interaction with Pasteuria ramosa makes them an excellent model system to investigate the genetics of host resistance. D. magna hosts were found to be either resistant (complete lack of attachment of parasite spores to the host’s foregut) or susceptible (full attachment). In this study the authors carried out an elegant Mendelian genetic investigation by performing multiple crosses between four host genotypes differing in their resistance to two different parasite isolates [1]. Their results show that resistance of D. magna to each of the two P. ramosa isolates relies on Mendelian inheritance at two loci that are linked (A and B), each of them having two alleles with dominant resistance; furthermore resistance to one parasite isolate confers susceptibility to the other. They also show that a third locus appears to confer double resistance (C), but that even double resistant hosts remain susceptible to other parasite isolates, and hence that universal host resistance is lacking – all of this supporting the Red Queen theory. This paper demonstrates with a high level of clarity that host resistance is governed by multiple linked loci. The assumption of epistasis between resistance loci is supported, which makes it possible for sexual recombination to be maintained by antagonistic host-parasite coevolution. Reference [1] Metzger CMJA, Luijckx P, Bento G, Mariadassou M, Ebert D. 2016. The Red Queen lives: epistasis between linked resistance loci. Evolution 70:480-487. doi: 10.1111/evo.12854 | The Red Queen lives: epistasis between linked resistance loci | Metzger CMJA, Luijckx P, Bento G, Mariadassou M, Ebert D. | A popular theory explaining the maintenance of genetic recombination (sex) is the Red Queen Theory. This theory revolves around the idea that time-lagged negative frequency-dependent selection by parasites favors rare host genotypes generated thro... | | Evolutionary Dynamics, Evolutionary Theory, Reproduction and Sex, Species interactions | Adele Mennerat | 2016-12-14 13:58:53 | ||
05 Feb 2019
The quiescent X, the replicative Y and the AutosomesReplication-independent mutations: a universal signature ?Recommended by Nicolas Galtier based on reviews by Marc Robinson-Rechavi and Robert LanfearMutations are the primary source of genetic variation, and there is an obvious interest in characterizing and understanding the processes by which they appear. One particularly important question is the relative abundance, and nature, of replication-dependent and replication-independent mutations - the former arise as cells replicate due to DNA polymerization errors, whereas the latter are unrelated to the cell cycle. A recent experimental study in fission yeast identified a signature of mutations in quiescent (=non-replicating) cells: the spectrum of such mutations is characterized by an enrichment in insertions and deletions (indels) compared to point mutations, and an enrichment of deletions compared to insertions [2]. References [1] Achaz, G., Gangloff, S., and Arcangioli, B. (2019). The quiescent X, the replicative Y and the Autosomes. BioRxiv, 351288, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/351288 | The quiescent X, the replicative Y and the Autosomes | Guillaume Achaz, Serge Gangloff, Benoit Arcangioli | <p>From the analysis of the mutation spectrum in the 2,504 sequenced human genomes from the 1000 genomes project (phase 3), we show that sexual chromosomes (X and Y) exhibit a different proportion of indel mutations than autosomes (A), ranking the... | | Bioinformatics & Computational Biology, Genome Evolution, Human Evolution, Molecular Evolution, Population Genetics / Genomics, Reproduction and Sex | Nicolas Galtier | 2018-07-25 10:37:48 | ||
30 Aug 2021
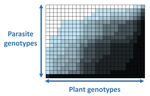
The quasi-universality of nestedness in the structure of quantitative plant-parasite interactionsNestedness and modularity in plant-parasite infection networksRecommended by Santiago Elena based on reviews by Rubén González and 2 anonymous reviewers based on reviews by Rubén González and 2 anonymous reviewers
In a landmark paper, Flores et al. (2011) showed that the interactions between bacteria and their viruses could be nicely described using a bipartite infection networks. Two quantitative properties of these networks were of particular interest, namely modularity and nestedness. Modularity emerges when groups of host species (or genotypes) shared groups of viruses. Nestedness provided a view of the degree of specialization of both partners: high nestedness suggests that hosts differ in their susceptibility to infection, with some highly susceptible host genotypes selecting for very specialized viruses while strongly resistant host genotypes select for generalist viruses. Translated to the plant pathology parlance, this extreme case would be equivalent to a gene-for-gene infection model (Flor 1956): new mutations confer hosts with resistance to recently evolved viruses while maintaining resistance to past viruses. Likewise, virus mutations for expanding host range evolve without losing the ability to infect ancestral host genotypes. By contrast, a non-nested network would represent a matching-allele infection model (Frank 2000) in which each interacting organism evolves by losing its capacity to resist/infect its ancestral partners, resembling a Red Queen dynamic. Obviously, the reality is more complex and may lie anywhere between these two extreme situations. Recently, Valverde et al. (2020) developed a model to explain the emergence of nestedness and modularity in plant-virus infection networks across diverse habitats. They found that local modularity could coexist with global nestedness and that intraspecific competition was the main driver of the evolution of ecosystems in a continuum between nested-modular and nested networks. These predictions were tested with field data showing the association between plant host species and different viruses in different agroecosystems (Valverde et al. 2020). The effect of interspecific competition in the structure of empirical plant host-virus infection networks was also tested by McLeish et al. (2019). Besides data from agroecosystems, evolution experiments have also shown the pervasive emergence of nestedness during the diversification of independently-evolved lineages of potyviruses in Arabidopsis thaliana genotypes that differ in their susceptibility to infection (Hillung et al. 2014; González et al. 2019; Navarro et al. 2020). In their study, Moury et al. (2021) have expanded all these previous observations to a diverse set of pathosystems that range from viruses, bacteria, oomycetes, fungi, nematodes to insects. While modularity was barely seen in only a few of the systems, nestedness was a common trend (observed in ~94% of all systems). This nestedness, as seen in previous studies and as predicted by theory, emerged as a consequence of the existence of generalist and specialist strains of the parasites that differed in their capacity to infect more or less resistant plant genotypes. As pointed out by Moury et al. (2021) in their conclusions, the ubiquity of nestedness in plant-parasite infection matrices has strong implications for the evolution and management of infectious diseases. References Flor, H. H. (1956). The complementary genic systems in flax and flax rust. In Advances in genetics, 8, 29-54. https://doi.org/10.1016/S0065-2660(08)60498-8 Flores, C. O., Meyer, J. R., Valverde, S., Farr, L., and Weitz, J. S. (2011). Statistical structure of host–phage interactions. Proceedings of the National Academy of Sciences, 108, E288-E297. https://doi.org/10.1073/pnas.1101595108 Frank, S. A. (2000). Specific and non-specific defense against parasitic attack. Journal of Theoretical Biology, 202, 283-304. https://doi.org/10.1006/jtbi.1999.1054 González, R., Butković, A., and Elena, S. F. (2019). Role of host genetic diversity for susceptibility-to-infection in the evolution of virulence of a plant virus. Virus evolution, 5(2), vez024. https://doi.org/10.1093/ve/vez052 Hillung, J., Cuevas, J. M., Valverde, S., and Elena, S. F. (2014). Experimental evolution of an emerging plant virus in host genotypes that differ in their susceptibility to infection. Evolution, 68, 2467-2480. https://doi.org/10.1111/evo.12458 McLeish, M., Sacristán, S., Fraile, A., and García-Arenal, F. (2019). Coinfection organizes epidemiological networks of viruses and hosts and reveals hubs of transmission. Phytopathology, 109, 1003-1010. https://doi.org/10.1094/PHYTO-08-18-0293-R Moury B, Audergon J-M, Baudracco-Arnas S, Krima SB, Bertrand F, Boissot N, Buisson M, Caffier V, Cantet M, Chanéac S, Constant C, Delmotte F, Dogimont C, Doumayrou J, Fabre F, Fournet S, Grimault V, Jaunet T, Justafré I, Lefebvre V, Losdat D, Marcel TC, Montarry J, Morris CE, Omrani M, Paineau M, Perrot S, Pilet-Nayel M-L and Ruellan Y (2021) The quasi-universality of nestedness in the structure of quantitative plant-parasite interactions. bioRxiv, 2021.03.03.433745, ver. 4 recommended and peer-reviewed by PCI Evolutionary Biology. https://doi.org/10.1101/2021.03.03.433745 Navarro, R., Ambros, S., Martinez, F., Wu, B., Carrasco, J. L., and Elena, S. F. (2020). Defects in plant immunity modulate the rates and patterns of RNA virus evolution. bioRxiv. doi: https://doi.org/10.1101/2020.10.13.337402 Valverde, S., Vidiella, B., Montañez, R., Fraile, A., Sacristán, S., and García-Arenal, F. (2020). Coexistence of nestedness and modularity in host–pathogen infection networks. Nature ecology & evolution, 4, 568-577. https://doi.org/10.1038/s41559-020-1130-9 | The quasi-universality of nestedness in the structure of quantitative plant-parasite interactions | Moury Benoît, Audergon Jean-Marc, Baudracco-Arnas Sylvie, Ben Krima Safa, Bertrand François, Boissot Nathalie, Buisson Mireille, Caffier Valérie, Cantet Mélissa, Chanéac Sylvia, Constant Carole, Delmotte François, Dogimont Catherine, Doumayrou Jul... | <p>Understanding the relationships between host range and pathogenicity for parasites, and between the efficiency and scope of immunity for hosts are essential to implement efficient disease control strategies. In the case of plant parasites, most... | | Bioinformatics & Computational Biology, Evolutionary Dynamics, Species interactions | Santiago Elena | 2021-03-04 21:23:08 | ||
13 Apr 2023
The landscape of nucleotide diversity in Drosophila melanogaster is shaped by mutation rate variationAn unusual suspect: the mutation landscape as a determinant of local variation in nucleotide diversityRecommended by Fernando Racimo based on reviews by David Castellano and 1 anonymous reviewerSometimes, important factors for explaining biological processes fall through the cracks, and it is only through careful modeling that their importance eventually comes out to light. In this study, Barroso and Dutheil introduce a new method based on the sequentially Markovian coalescent (SMC, Marjoran and Wall 2006) for jointly estimating local recombination and coalescent rates along a genome. Unlike previous SMC-based methods, however, their method can also co-estimate local patterns of variation in mutation rates. This is a powerful improvement which allows them to tackle questions about the reasons for the extensive variation in nucleotide diversity across the chromosomes of a species - a problem that has plagued the minds of population geneticists for decades (Begun and Aquadro 1992, Andolfatto 2007, McVicker et al., 2009, Pouyet and Gilbert 2021). The authors find that variation in de novo mutation rates appears to be the most important factor in determining nucleotide diversity in Drosophila melanogaster. Though seemingly contradicting previous attempts at addressing this problem (Comeron 2014), they take care to investigate and explain why that might be the case. Barroso and Dutheil have also taken care to carefully explain the details of their new approach and have carried a very thorough set of analyses comparing competing explanations for patterns of nucleotide variation via causal modeling. The reviewers raised several issues involving choices made by the authors in their analysis of variance partitioning, the proper evaluation of the role of linked selection and the recombination rate estimates emerging from their model. These issues have all been extensively addressed by the authors, and their conclusions seem to remain robust. The study illustrates why the mutation landscape should not be ignored as an important determinant of local variation in genetic diversity, and opens up questions about the generalizability of these results to other organisms. REFERENCES Andolfatto, P. (2007). Hitchhiking effects of recurrent beneficial amino acid substitutions in the Drosophila melanogaster genome. Genome research, 17(12), 1755-1762. https://doi.org/10.1101/gr.6691007 Barroso, G. V., & Dutheil, J. Y. (2021). The landscape of nucleotide diversity in Drosophila melanogaster is shaped by mutation rate variation. bioRxiv, 2021.09.16.460667, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.09.16.460667 Begun, D. J., & Aquadro, C. F. (1992). Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature, 356(6369), 519-520. https://doi.org/10.1038/356519a0 Comeron, J. M. (2014). Background selection as baseline for nucleotide variation across the Drosophila genome. PLoS Genetics, 10(6), e1004434. https://doi.org/10.1371/journal.pgen.1004434 Marjoram, P., & Wall, J. D. (2006). Fast" coalescent" simulation. BMC genetics, 7, 1-9. https://doi.org/10.1186/1471-2156-7-16 McVicker, G., Gordon, D., Davis, C., & Green, P. (2009). Widespread genomic signatures of natural selection in hominid evolution. PLoS genetics, 5(5), e1000471. https://doi.org/10.1371/journal.pgen.1000471 Pouyet, F., & Gilbert, K. J. (2021). Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between. Peer Community Journal, 1, e27. https://doi.org/10.24072/pcjournal.16 | The landscape of nucleotide diversity in Drosophila melanogaster is shaped by mutation rate variation | Gustavo V Barroso, Julien Y Dutheil | <p style="text-align: justify;">What shapes the distribution of nucleotide diversity along the genome? Attempts to answer this question have sparked debate about the roles of neutral stochastic processes and natural selection in molecular evolutio... | | Bioinformatics & Computational Biology, Population Genetics / Genomics | Fernando Racimo | 2022-10-30 07:52:07 | ||
25 Mar 2019

The joint evolution of lifespan and self-fertilisationEvolution of selfing & lifespan 2.0Recommended by Thomas Bataillon based on reviews by 2 anonymous reviewersFlowering plants display a staggering diversity of both mating systems and life histories, ranging from almost exclusively selfers to obligate outcrossers, very short-lived annual herbs to super long lived trees. One pervasive pattern that has attracted considerable attention is the tight correlation that is found between mating systems and lifespan [1]. Until recently, theoretical explanations for these patterns have relied on static models exploring the consequences of several non-mutually exclusive important process: levels of inbreeding depression and ability to successfully were center stage. This make sense: successful colonization after long‐distance dispersal is far more likely to happen for self‐compatible than for self‐incompatible individuals in a sexually reproducing species. Furthermore, inbreeding depression (essentially a genetically driven phenomenon) and reproductive insurance are expected to shape the evolution of both mating system and lifespan. References | The joint evolution of lifespan and self-fertilisation | Thomas Lesaffre, Sylvain Billiard | <p>In Angiosperms, there exists a strong association between mating system and lifespan. Most self-fertilising species are short-lived and most predominant or obligate outcrossers are long-lived. This association is generally explained by the infl... | | Evolutionary Theory, Life History, Reproduction and Sex | Thomas Bataillon | 2018-09-19 10:03:51 | ||
18 May 2020
The insertion of a mitochondrial selfish element into the nuclear genome and its consequencesSome evolutionary insights into an accidental homing endonuclease passage from mitochondria to the nucleusRecommended by Sylvain Charlat based on reviews by Jan Engelstaedter and Yannick WurmNot all genetic elements composing genomes are there for the benefit of their carrier. Many have no consequences on fitness, or too mild ones to be eliminated by selection, and thus stem from neutral processes. Many others are indeed the product of selection, but one acting at a different level, increasing the fitness of some elements of the genome only, at the expense of the “organism” as a whole. These can be called selfish genetic elements, and come into a wide variety of flavours [1], illustrating many possible means to cheat with “fair” reproductive processes such as meiosis, and thus get overrepresented in the offspring of their hosts. Producing copies of itself through transposition is one such strategy; a very successful one indeed, explaining a large part of the genomic content of many organisms. Killing non carrier gametes following meiosis in heterozygous carriers is another one. Less know and less common is the ability of some elements to turn heterozygous carriers into homozygous ones, that will thus transmit the selfish elements to all offspring instead of half. This is achieved by nucleic sequences encoding so-called “Homing endonucleases” (HEs). These proteins tend to induce double strand breaks of DNA specifically in regions homologous to their own insertion sites. The recombination machinery is such that the intact homologous region, that is, the one carrying the HE sequence, is then used as a template for the reparation of the break, resulting in the effective conversion of a non-carrier allele into a carrier allele. Such elements can also occur in the mitochondrial genomes of organisms where mitochondria are not strictly transmitted by one parent only, offering mitochondrial HEs some opportunities for “homing” into new non carrier genomes. This is the case in yeasts, where HEs were first reported [2,3]. References [1] Burt, A., and Trivers, R. (2006). Genes in Conflict: The Biology of Selfish Genetic Elements. Belknap Press. | The insertion of a mitochondrial selfish element into the nuclear genome and its consequences | Julien Y. Dutheil, Karin Münch, Klaas Schotanus, Eva H. Stukenbrock and Regine Kahmann | <p>Homing endonucleases (HE) are enzymes capable of cutting DNA at highly specific target sequences, the repair of the generated double-strand break resulting in the insertion of the HE-encoding gene ("homing" mechanism). HEs are present in all th... | | Genome Evolution, Molecular Evolution | Sylvain Charlat | 2019-09-30 20:34:23 | ||
23 Jan 2023
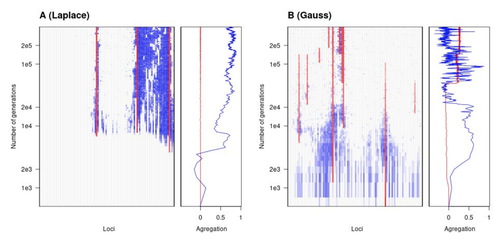
The genetic architecture of local adaptation in a clineEnvironmental and fitness landscapes matter for the genetic basis of local adaptationRecommended by Charles Mullon based on reviews by 2 anonymous reviewers
Natural landscapes are often composite, with spatial variation in environmental factors being the norm rather than exception. Adaptation to such variation is a major driver of diversity at all levels of biological organization, from genes to phenotypes, species and ultimately ecosystems. While natural selection favours traits that show a better fit to local conditions, the genomic response to such selection is not necessarily straightforward. This is because many quantitative traits are complex and the product of many loci, each with a small to moderate phenotypic contribution. Adapting to environmental challenges that occur in narrow ranges may thus prove difficult as each individual locus is easily swamped by alleles favoured across the rest of the population range. To better understand whether and how evolution overcomes such a hurdle, Laroche and Lenormand [1] combine quantitative genetics and population genetic modelling to track genomic changes that underpin a trait whose fitness optimum differs between a certain spatial range, referred to as a “pocket”, and the rest of the habitat. As it turns out from their analysis, one critical and probably underappreciated factor in determining the type of genetic architecture that evolves is how fitness declines away from phenotypic optima. One classical and popular model of fitness landscape that relates trait value to reproductive success is Gaussian, whereby small trait variations away from the optimum result in even smaller variations in fitness. This facilitates local adaptation via the invasion of alleles of small effects as carriers inside the pocket show a better fit while those outside the pocket only suffer a weak fitness cost. By contrast, when the fitness landscape is more peaked around the optimum, for instance where the decline is linear, adaptation through weak effect alleles is less likely, requiring larger pockets that are less easily swamped by alleles selected in the rest of the range. In addition to mathematically investigating the initial emergence of local adaptation, Laroche and Lenormand use computer simulations to look at its long-term maintenance. In principle, selection should favour a genetic architecture that consolidates the phenotype and increases its heritability, for instance by grouping several alleles of large effects close to one another on a chromosome to avoid being broken down by meiotic recombination. Whether or not this occurs also depends on the fitness landscape. When the landscape is Gaussian, the genetic architecture of the trait eventually consists of tightly linked alleles of large effects. The replacement of small effects by large effects loci is here again promoted by the slow fitness decline around the optimum. This is because any shift in architecture in an adapted population requires initially crossing a fitness valley. With a Gaussian landscape, this valley is shallow enough to be crossed, facilitated by a bit of genetic drift. By contrast, when fitness declines linearly around the optimum, genetic architecture is much less evolutionarily labile as any architecture change initially entails a fitness cost that is too high to bear. Overall, Laroche and Lenormand provide a careful and thought-provoking analysis of a classical problem in population genetics. In addition to questioning some longstanding modelling assumptions, their results may help understand why differentiated populations are sometimes characterized by “genomic islands” of divergence, and sometimes not. References [1] Laroche F, Lenormand T (2022) The genetic architecture of local adaptation in a cline. bioRxiv, 2022.06.30.498280, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.06.30.498280 | The genetic architecture of local adaptation in a cline | Fabien Laroche, Thomas Lenormand | <p>Local adaptation is pervasive. It occurs whenever selection favors different phenotypes in different environments, provided that there is genetic variation for the corresponding traits and that the effect of selection is greater than the effect... | | Adaptation, Evolutionary Theory, Genome Evolution, Molecular Evolution, Population Genetics / Genomics, Quantitative Genetics | Charles Mullon | 2022-07-07 08:46:47 | ||
18 Jan 2023
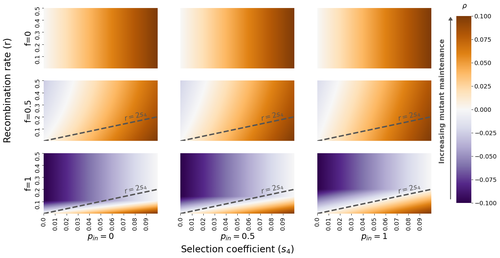
The fate of recessive deleterious or overdominant mutations near mating-type loci under partial selfingMaintenance of deleterious mutations and recombination suppression near mating-type loci under selfingRecommended by Aurelien Tellier based on reviews by 3 anonymous reviewersThe causes and consequences of the evolution of sexual reproduction are a major topic in evolutionary biology. With advances in sequencing technology, it becomes possible to compare sexual chromosomes across species and infer the neutral and selective processes shaping polymorphism at these chromosomes. Most sex and mating-type chromosomes exhibit an absence of recombination in large genomic regions around the animal, plant or fungal sex-determining genes. This suppression of recombination likely occurred in several time steps generating stepwise increasing genomic regions starting around the sex-determining genes. This mechanism generates so-called evolutionary strata of differentiation between sex chromosomes (Nicolas et al., 2004, Bergero and Charlesworth, 2009, Hartmann et al. 2021). The evolution of extended regions of recombination suppression is also documented on mating-type chromosomes in fungi (Hartmann et al., 2021) and around supergenes (Yan et al., 2020, Jay et al., 2021). The exact reason and evolutionary mechanisms for this phenomenon are still, however, debated. Two hypotheses are proposed: 1) sexual antagonism (Charlesworth et al., 2005), which, nevertheless, does explain the observed occurrence of the evolutionary strata, and 2) the sheltering of deleterious alleles by inversions carrying a lower load than average in the population (Charlesworth and Wall, 1999, Antonovics and Abrams, 2004). In the latter, the mechanism is as follows. A genetic inversion or a suppressor of recombination in cis may exhibit some overdominance behaviour. The inversion exhibiting less recessive deleterious mutations (compared to others at the same locus) may increase in frequency, before at higher frequency occurring at the homozygous state, expressing its genetic load. However, the inversion may never be at the homozygous state if it is genetically linked to a gene in a permanently heterozygous state. The inversion can then be advantageous and may reach fixation at the sex chromosome (Charlesworth and Wall, 1999, Antonovics and Abrams, 2004, Jay et al., 2022). These selective mechanisms promote thus the suppression of recombination around the sex-determining gene, and recessive deleterious mutations are permanently sheltered. This hypothesis is corroborated by the sheltering of deleterious mutations observed around loci under balancing selection (Llaurens et al. 2009, Lenz et al. 2016) and around mating-type genes in fungi and supergenes (Jay et al. 2021, Jay et al., 2022). In this present theoretical study, Tezenas et al. (2022) analyse how linkage to a necessarily heterozygous fungal mating type locus influences the persistence/extinction time of a new mutation at a second selected locus. This mutation can either be deleterious and recessive, or overdominant. There is arbitrary linkage between the two loci, and sexual reproduction occurs either between 1) gametes of different individuals (outcrossing), or 2) by selfing with gametes originating from the same (intra-tetrad) or different (inter-tetrad) tetrads produced by that individual. Note, here, that the mating-type gene does not prevent selfing. The authors study the initial stochastic dynamics of the mutation using a multi-type branching process (and simulations when analytical results cannot be obtained) to compute the extinction time of the deleterious mutation. The main result is that the presence of a mating-type locus always decreases the purging probability and increases the purging time of the mutations under selfing. Ultimately, deleterious mutations can indeed accumulate near the mating-type locus over evolutionary time scales. In a nutshell, high selfing or high intra-tetrad mating do increase the sheltering effect of the mating-type locus. In effect, the outcome of sheltering of deleterious mutations depends on two opposing mechanisms: 1) a higher selfing rate induces a greater production of homozygotes and an increased effect of the purging of deleterious mutations, while 2) a higher intra-tetrad selfing rate (or linkage with the mating-type locus) generates heterozygotes which have a small genetic load (and are favoured). The authors also show that rare events of extremely long maintenance of deleterious mutations can occur. The authors conclude by highlighting the manifold effect of selfing which reduces the effective population size and thus impairs the efficiency of selection and increases the mutational load, while also favouring the purge of deleterious homozygous mutations. Furthermore, this study emphasizes the importance of studying the maintenance and accumulation of deleterious mutations in the vicinity of heterozygous loci (e.g. under balancing selection) in selfing species. References Antonovics J, Abrams JY (2004) Intratetrad Mating and the Evolution of Linkage Relationships. Evolution, 58, 702–709. https://doi.org/10.1111/j.0014-3820.2004.tb00403.x Bergero R, Charlesworth D (2009) The evolution of restricted recombination in sex chromosomes. Trends in Ecology & Evolution, 24, 94–102. https://doi.org/10.1016/j.tree.2008.09.010 Charlesworth D, Morgan MT, Charlesworth B (1990) Inbreeding Depression, Genetic Load, and the Evolution of Outcrossing Rates in a Multilocus System with No Linkage. Evolution, 44, 1469–1489. https://doi.org/10.1111/j.1558-5646.1990.tb03839.x Charlesworth D, Charlesworth B, Marais G (2005) Steps in the evolution of heteromorphic sex chromosomes. Heredity, 95, 118–128. https://doi.org/10.1038/sj.hdy.6800697 Charlesworth B, Wall JD (1999) Inbreeding, heterozygote advantage and the evolution of neo–X and neo–Y sex chromosomes. Proceedings of the Royal Society of London. Series B: Biological Sciences, 266, 51–56. https://doi.org/10.1098/rspb.1999.0603 Hartmann FE, Duhamel M, Carpentier F, Hood ME, Foulongne-Oriol M, Silar P, Malagnac F, Grognet P, Giraud T (2021) Recombination suppression and evolutionary strata around mating-type loci in fungi: documenting patterns and understanding evolutionary and mechanistic causes. New Phytologist, 229, 2470–2491. https://doi.org/10.1111/nph.17039 Jay P, Chouteau M, Whibley A, Bastide H, Parrinello H, Llaurens V, Joron M (2021) Mutation load at a mimicry supergene sheds new light on the evolution of inversion polymorphisms. Nature Genetics, 53, 288–293. https://doi.org/10.1038/s41588-020-00771-1 Jay P, Tezenas E, Véber A, Giraud T (2022) Sheltering of deleterious mutations explains the stepwise extension of recombination suppression on sex chromosomes and other supergenes. PLOS Biology, 20, e3001698. https://doi.org/10.1371/journal.pbio.3001698 Lenz TL, Spirin V, Jordan DM, Sunyaev SR (2016) Excess of Deleterious Mutations around HLA Genes Reveals Evolutionary Cost of Balancing Selection. Molecular Biology and Evolution, 33, 2555–2564. https://doi.org/10.1093/molbev/msw127 Llaurens V, Gonthier L, Billiard S (2009) The Sheltered Genetic Load Linked to the S Locus in Plants: New Insights From Theoretical and Empirical Approaches in Sporophytic Self-Incompatibility. Genetics, 183, 1105–1118. https://doi.org/10.1534/genetics.109.102707 Nicolas M, Marais G, Hykelova V, Janousek B, Laporte V, Vyskot B, Mouchiroud D, Negrutiu I, Charlesworth D, Monéger F (2004) A Gradual Process of Recombination Restriction in the Evolutionary History of the Sex Chromosomes in Dioecious Plants. PLOS Biology, 3, e4. https://doi.org/10.1371/journal.pbio.0030004 Tezenas E, Giraud T, Véber A, Billiard S (2022) The fate of recessive deleterious or overdominant mutations near mating-type loci under partial selfing. bioRxiv, 2022.10.07.511119, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.10.07.511119 Yan Z, Martin SH, Gotzek D, Arsenault SV, Duchen P, Helleu Q, Riba-Grognuz O, Hunt BG, Salamin N, Shoemaker D, Ross KG, Keller L (2020) Evolution of a supergene that regulates a trans-species social polymorphism. Nature Ecology & Evolution, 4, 240–249. https://doi.org/10.1038/s41559-019-1081-1 | The fate of recessive deleterious or overdominant mutations near mating-type loci under partial selfing | Emilie Tezenas, Tatiana Giraud, Amandine Veber, Sylvain Billiard | <p style="text-align: justify;">Large regions of suppressed recombination having extended over time occur in many organisms around genes involved in mating compatibility (sex-determining or mating-type genes). The sheltering of deleterious alleles... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Theory, Genome Evolution, Population Genetics / Genomics, Reproduction and Sex | Aurelien Tellier | 2022-10-10 13:50:30 | ||
03 Oct 2023
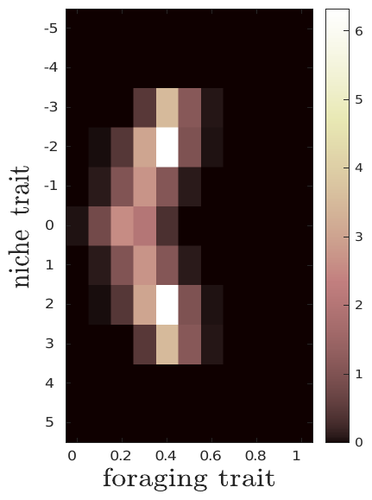
The evolutionary dynamics of plastic foraging and its ecological consequences: a resource-consumer modelEvolution and consequences of plastic foraging behavior in consumer-resource ecosystemsRecommended by François Rousset based on reviews by 2 anonymous reviewersPlastic responses of organisms to their environment may be maladaptive in particular when organisms are exposed to new environments. Phenotypic plasticity may also have opposite effects on the adaptive response of organisms to environmental changes: whether phenotypic plasticity favors or hinders such adaptation depends on a balance between the ability of the population to respond to the change non-genetically in the short term, and the weakened genetic response to environmental change. These topics have received continued attention, particularly in the context of climate change (e.g., Chevin et al. 2013, Duputié et al., 2015, Vinton et al . 2022). In their work, Ledru et al. focus on the adaptive nature of plastic behavior and on its consequences in a consumer-resource ecosystem. As they emphasize, previous works have found that plastic foraging promotes community stability, but these did not consider plasticity as an evolving trait, so Ledru et al. set out to test whether this conclusion holds when both plastic foraging and niche traits of consumers and resources evolve (though ultimately, their new conclusions may not all depend on plasticity evolving). Along the way, they first seek to clarify when such plasticity will evolve, and how it affects the evolution of the niche diversity of consumers and resources, before turning to the question of consumer persistence. The model is rather complex, as three traits are allowed to evolve, and the resource uptake expressed through plastic behavior has its own dynamics affected by some form of social learning. Classically, in models of niche evolution, a consumer's efficiency in exploiting a resource characterized by a trait y (here, the resource's individual niche trait), has been described in terms of location-scale (typically Gaussian) kernels, with mean x (the consumer's individual niche trait) specifying the most efficiently exploited resource, and with variance characterizing individual niche breadth. The evolution of the variance has been considered in some previous models but is assumed to be fixed here. Rather, the new model considers the evolution of the distribution of resource traits, of the consumer's individual niche trait (which is not plastic), and of a "plastic foraging trait" that controls the relative time spent foraging plastically versus foraging randomly. When foraging plastically, the consumers modify their foraging effort towards the type of resource that maximizes their energy intake. in some previous models, the effect of variation in the extent of plastic foraging was already considered, but the evolution of allocation to a plastic foraging strategy versus random foraging was not considered. The model is formulated through reaction-diffusion equations, and its dynamics is investigated by numerical integration. Foraging plasticity readily evolves, when resources vary widely enough, competition for resources is strong, and the cost of plasticity is weak. This means in particular that a large individual niche width of consumers selects for increased plastic foraging, as the evolution of plastic foraging leads to reduced niche overlap between consumers. The evolution of plastic foraging itself generally, though not always, favors the diversification of the niche traits of consumers and of resources. There is thus a positive feedback loop between plastic foraging and resource diversity. Ledru et al. conclude that the total niche width of the consumer population should also correlate with the evolution of plastic foraging, an implication which they relate to the so-called niche variation hypothesis and to empirical tests of it. The joint evolution of the consumer's individual niche trait and plastic foraging trait generates a striking pattern within populations: consumers whose individual niche trait is at an edge of the resource distribution forage more plastically. The authors observe that this relatively simple prediction has not been subjected to any empirical test. Returning to the question of consumer persistence, Ledru et al. evaluate this persistence when consumer mortality increases, and in response to either gradual or sudden environmental changes. These different perturbations all reduce the benefits of plastic foraging. The effect of plastic foraging on stability are complex, being negative or positive effect depending on the type of disturbance, and in particular the ecosystem has a lower sustainable rate of environmental change in the presence of plastic foraging. However, allowing the evolutionary regression of plastic foraging then has a comparatively positive effect on persistence. Despite the substantial effort devoted to analyzing this complex model, relaxing some of its assumptions would likely reveal further complexities. Notably, the overall effect of plasticity on consumer persistence depends on effects already encountered in models of the adaptive response of single species to environmental change: a fast non-genetic response in the short term versus a weakened genetic response in the longer term. The overall balance between these opposite effects on adaptation may be difficult to predict robustly. In the case of a constant rate of environmental change, the results of the present model depend on a lag load between the trait changes of consumer and resource populations, and the extent of the lag may also depend on many factors, such as the extent of genetic variation (e.g., Bürger & Lynch, 1995) for niche traits in consumers and resources. Here, the same variance of mutational effects was assumed for all three evolving traits. Further, spatial environmental variation, a central issue in studies of adaptive responses to environmental changes (e.g., Parmesan, 2006, Zhu et al., 2012), was not considered. Finally, the rate of adjustment of effort by consumers with given niche trait and plastic foraging trait values was assumed proportional to the density of consumers with such trait values. This was justified as a way of accounting for the use of social cues during foraging, but to the extent that they occur, social effects could manifest themselves through other learning dynamics. In conclusion, Ledru et al. have addressed a broad range of questions, suggesting new empirical tests of behavioural patterns on one side, and recovering in the context of community response to environmental changes a complexity that could be expected from earlier works on adaptive responses of organisms but that has been overlooked by previous works on community effects of phenotypic plasticity. References Bürger, R. and Lynch, M. (1995), Evolution and extinction in a changing environment: a quantitative-genetic analysis. Evolution, 49: 151-163. https://doi.org/10.1111/j.1558-5646.1995.tb05967.x Chevin, L.-M., Collins, S. and Lefèvre, F. (2013), Phenotypic plasticity and evolutionary demographic responses to climate change: taking theory out to the field. Funct Ecol, 27: 967-979. https://doi.org/10.1111/j.1365-2435.2012.02043.x Duputié, A., Rutschmann, A., Ronce, O. and Chuine, I. (2015), Phenological plasticity will not help all species adapt to climate change. Glob Change Biol, 21: 3062-3073. https://doi-org.inee.bib.cnrs.fr/10.1111/gcb.12914 Ledru, L., Garnier, J., Guillot, O., Faou, E., & Ibanez, S. (2023). The evolutionary dynamics of plastic foraging and its ecological consequences: a resource-consumer model. EcoEvoRxiv, ver. 4 peer-reviewed and recommended by Peer Community In Evolutionary Biology. https://doi.org/10.32942/X2QG7M Parmesan, C. (2006) Ecological and evolutionary responses to recent climate change Vinton, A.C., Gascoigne, S.J.L., Sepil, I., Salguero-Gómez, R., (2022) Plasticity’s role in adaptive evolution depends on environmental change components. Trends in Ecology & Evolution, 37: 1067-1078. Zhu, K., Woodall, C.W. and Clark, J.S. (2012), Failure to migrate: lack of tree range expansion in response to climate change. Glob Change Biol, 18: 1042-1052. https://doi.org/10.1111/j.1365-2486.2011.02571.x | The evolutionary dynamics of plastic foraging and its ecological consequences: a resource-consumer model | Léo Ledru, Jimmy Garnier, Océane Guillot, Erwan Faou, Camille Noûs, Sébastien Ibanez | <p style="text-align: justify;">Phenotypic plasticity has important ecological and evolutionary consequences. In particular, behavioural phenotypic plasticity such as plastic foraging (PF) by consumers, may enhance community stability. Yet little ... | | Bioinformatics & Computational Biology, Evolutionary Dynamics, Evolutionary Ecology, Phenotypic Plasticity | François Rousset | 2023-03-25 12:04:08 |




