
Latest recommendations

| Id | Title * | Authors * | Abstract * ▲ | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
16 Dec 2016
POSTPRINT
Spatiotemporal microbial evolution on antibiotic landscapesA poster child for experimental evolutionRecommended by Daniel Rozen and Arjan de VisserEvolution is usually studied via two distinct approaches: by inferring evolutionary processes from relatedness patterns among living species or by observing evolution in action in the laboratory or field. A recent study by Baym and colleagues in Science [1] has now combined these approaches by taking advantage of the pattern left behind by spatially evolving bacterial populations. Evolution is often considered too slow to see, and can only be inferred by studying patterns of relatedness using phylogenetic trees. Increasingly, however, researchers are moving nature into the lab and watching as evolution unfolds under their noses. The field of experimental evolution follows evolutionary change in the laboratory over 10s to 1000s of generations, yielding insights into bacterial, viral, plant, or fly evolution (among many other species) that are simply not possible in the field. Yet, as powerful as experimental evolution is, it lacks a posterchild. There is no Galapagos finch radiation, nor a stunning series of cichlids to showcase to our students to pique their interests. Let’s face it, E. coli is no stickleback! And while practitioners of experimental evolution can explain the virtues of examining 60,000 generations of bacterial evolution in action, appreciating this nevertheless requires a level of insight and imagination that often eludes students, who need to see “it” to get it. Enter MEGA, an idea and a film that could become the new face of experimental evolution. It replaces big numbers of generations or images of scientists, with an actual picture of the scientific result. MEGA, or Microbial Evolution and Growth Arena, is essentially an enormous petri dish and is the brainchild of Michael Baym, Tami Leiberman and their colleagues in Roy Kishony’s lab at Technion Israel Institute of Technology and Harvard Medical School. The idea of MEGA is to allow bacteria to swim over a spatially defined landscape while adapting to the local conditions, in this case antibiotics. When bacteria are inoculated onto one end of the plate they consume resources while swarming forward from the plate edge. In a few days, the bacteria grow into an area with antibiotics to which they are susceptible. This stops growth until a mutation arises that permits the bacteria to jump this hurdle, after which growth proceeds until the next hurdle of a 10-fold higher antibiotic concentration, and so on. By this simple approach, Baym et al. [1] evolved E. coli that were nearly 105-fold more resistant to two different antibiotics in just over 10 days. In addition, they identified the mutations that were required for these changes, showed that mutations conferring smaller benefits were required before bacteria could evolve maximal resistance, observed changes to the mutation rate, and demonstrated the importance of spatial structure in constraining adaptation. For one thing, the rate of resistance evolution is impressive, and also quite scary given the mounting threat of antibiotic-resistant pathogens. However, MEGA also offers a uniquely visual insight into evolutionary change. By taking successive images of the MEGA plate, the group was able to watch the bacteria move, get trapped because of their susceptibility to the antibiotic, and then get past these traps as new mutations emerged that increased resistance. Each transition showcases evolution in real time. In addition, by leaving a spatial pattern of evolutionary steps behind, the MEGA plate offers unique opportunities to thoroughly investigate these steps when the experiment is finished. For instance, subsequent steps in mutational pathways can be characterized, but also their effects on fitness can be quantified in situ by measuring changes in survival and reproduction. This new method is undoubtedly a boon to the field of experimental evolution and offers endless opportunities for experimental elaboration. Perhaps of equal importance, MEGA is a tool that is portable to the classroom and to the public at large. Don’t believe in evolution? Watch this. You only have time for a short internship or lab practical? No problem. Don’t worry much about antibiotic resistance? Check this out. Like the best experimental tools, MEGA is simple but allows for complicated insights. And even if it is less charismatic than a finch, it still allows for the kinds of “gee-whiz” insights that will get students hooked on evolutionary biology. Reference [1] Baym M, Lieberman TD, Kelsic ED, Chait R, Gross R, Yelin I, Kishony R. 2016. Spatiotemporal microbial evolution on antibiotic landscapes. Science 353:1147-1151. doi: 10.1126/science.aag0822 | Spatiotemporal microbial evolution on antibiotic landscapes | Baym M, Lieberman TD, Kelsic ED, Chait R, Gross R, Yelin I, Kishony R | A key aspect of bacterial survival is the ability to evolve while migrating across spatially varying environmental challenges. Laboratory experiments, however, often study evolution in well-mixed systems. Here, we introduce an experimental device,... | | Adaptation, Evolutionary Applications, Experimental Evolution | Daniel Rozen | 2016-12-14 14:26:06 | ||
14 Dec 2016

POSTPRINT
The Red Queen lives: epistasis between linked resistance lociEvidence of epistasis provides further support to the Red Queen theory of host-parasite coevolutionRecommended by Adele Mennerat and Thierry LefèvreAccording to the Red Queen theory of antagonistic host-parasite coevolution, adaptation of parasites to the most common host genotype results in negative frequency-dependent selection whereby rare host genotypes are favoured. Assuming that host resistance relies on a genetic host-parasite (mis)match involving several linked loci, then recombination appears as much more efficient than parthenogenesis in generating new resistant host genotypes. This has long been proposed to explain one of the biggest so-called paradoxes in evolutionary biology, i.e. the maintenance of recombination despite its twofold cost. Evidence from various study systems indicates that successful infection (and hence host resistance) depends on a genetic match between the parasite’s and the host’s genotype via molecular interactions involving elicitor/receptor mechanisms. However the key assumption of epistasis, i.e. that this genetic host-parasite match involves several linked resistance loci, remained unsupported so far. Metzger and coauthors [1] now provide empirical support for it. Daphnia magna can reproduce both sexually and clonally and their well-studied interaction with Pasteuria ramosa makes them an excellent model system to investigate the genetics of host resistance. D. magna hosts were found to be either resistant (complete lack of attachment of parasite spores to the host’s foregut) or susceptible (full attachment). In this study the authors carried out an elegant Mendelian genetic investigation by performing multiple crosses between four host genotypes differing in their resistance to two different parasite isolates [1]. Their results show that resistance of D. magna to each of the two P. ramosa isolates relies on Mendelian inheritance at two loci that are linked (A and B), each of them having two alleles with dominant resistance; furthermore resistance to one parasite isolate confers susceptibility to the other. They also show that a third locus appears to confer double resistance (C), but that even double resistant hosts remain susceptible to other parasite isolates, and hence that universal host resistance is lacking – all of this supporting the Red Queen theory. This paper demonstrates with a high level of clarity that host resistance is governed by multiple linked loci. The assumption of epistasis between resistance loci is supported, which makes it possible for sexual recombination to be maintained by antagonistic host-parasite coevolution. Reference [1] Metzger CMJA, Luijckx P, Bento G, Mariadassou M, Ebert D. 2016. The Red Queen lives: epistasis between linked resistance loci. Evolution 70:480-487. doi: 10.1111/evo.12854 | The Red Queen lives: epistasis between linked resistance loci | Metzger CMJA, Luijckx P, Bento G, Mariadassou M, Ebert D. | A popular theory explaining the maintenance of genetic recombination (sex) is the Red Queen Theory. This theory revolves around the idea that time-lagged negative frequency-dependent selection by parasites favors rare host genotypes generated thro... | | Evolutionary Dynamics, Evolutionary Theory, Reproduction and Sex, Species interactions | Adele Mennerat | 2016-12-14 13:58:53 | ||
13 Dec 2016
POSTPRINT
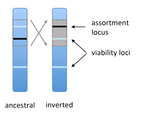
Prezygotic isolation, mating preferences, and the evolution of chromosomal inversionsThe spread of chromosomal inversions as a mechanism for reinforcementRecommended by Denis Roze and Thomas Broquet
Several examples of chromosomal inversions carrying genes affecting mate choice have been reported from various organisms. Furthermore, inversions are also frequently involved in genetic isolation between populations or species. Past work has shown that inversions can spread when they capture not only some loci involved in mate choice but also loci involved in incompatibilities between hybridizing populations [1]. In this new paper [2], the authors derive analytical approximations for the selection coefficient associated with an inversion suppressing recombination between a locus involved in mate choice and one (or several) locus involved in Dobzhansky-Muller incompatibilities. Two mechanisms for mate choice are considered: assortative mating based on the allele present at a single locus, or a trait-preference model where one locus codes for the trait and another for the preference. The results show that such an inversion is generally favoured, the selective advantage associated with the inversion being strongest when hybridization is sufficiently frequent. Assuming pairwise epistatic interactions between loci involved in incompatibilities, selection for the inversion increases approximately linearly with the number of such loci captured by the inversion. This paper is a good read for several reasons. First, it presents the problem clearly (e.g. the introduction provides a clear and concise presentation of the issue and past work) and its crystal-clear writing facilitates the reader's understanding of theoretical approaches and results. Second, the analysis is competently done and adds to previous work by showing that very general conditions are expected to be favourable to the spread of the type of inversion considered here. And third, it provides food for thought about the role of inversions in the origin or the reinforcement of divergence between nascent species. One result of this work is that an inversion linked to pre-zygotic isolation "is favoured so long as there is viability selection against recombinant genotypes", suggesting that genetic incompatibilities must have evolved first and that inversions capturing mating preference loci may then enhance pre-existing reproductive isolation. However, the results also show that inversions are more likely to be favoured in hybridizing populations among which gene flow is still high, rather than in more strongly isolated populations. This matches the observation that inversions are more frequently observed between sympatric species than between allopatric ones. References [1] Trickett AJ, Butlin RK. 1994. Recombination Suppressors and the Evolution of New Species. Heredity 73:339-345. doi: 10.1038/hdy.1994.180 [2] Dagilis AJ, Kirkpatrick M. 2016. Prezygotic isolation, mating preferences, and the evolution of chromosomal inversions. Evolution 70: 1465–1472. doi: 10.1111/evo.12954 | Prezygotic isolation, mating preferences, and the evolution of chromosomal inversions | Dagilis AJ, Kirkpatrick M | Chromosomal inversions are frequently implicated in isolating species. Models have shown how inversions can evolve in the context of postmating isolation. Inversions are also frequently associated with mating preferences, a topic that has not been... | | Adaptation, Evolutionary Theory, Genome Evolution, Hybridization / Introgression, Population Genetics / Genomics, Speciation | Denis Roze | 2016-12-13 22:11:54 | ||
15 Dec 2016

POSTPRINT
Limiting opportunities for cheating stabilizes virulence in insect parasitic nematodesApplication of kin theory to long-standing problem in nematode production for biocontrolRecommended by Thomas Sappington and Ruth Arabelle HufbauerMuch research effort has been extended toward developing systems for managing soil inhabiting insect pests of crops with entomopathogenic nematodes as biocontrol agents. Although small plot or laboratory experiments may suggest a particular insect pest is vulnerable to management in this way, it is often difficult to scale-up nematode production for application at the field- and farm scale to make such a tactic viable. Part of the problem is that entomopathogenic nematode strains must be propagated by serial passage in vivo, because storage by freezing decreases fitness. At the same time, serial propagation results in loss of virulence (ability to infect) over generations in the laboratory, a phenomenon called attenuation. To probe the underlying reasons for development of attenuation, as a prerequisite to designing strategies to mitigate it, Shapiro-Ilan and Raymond [1] turned to evolutionary theory of social conflict as a possible explanatory framework. Virulence of entomopathogenic nematodes depends on a combination of virulence factors, like various proteases, secreted by both the nematode and symbiotic bacteria to overcome host defenses. Attenuation is characterized in part by a reduced production of these factors. Invasion of a host involves simultaneous attack by a group of nematodes ("cooperators"), which together neutralize host defenses enough to allow individuals to successfully invade. "Cheaters" in the invading population can avoid the metabolic costs of producing virulence factors while reaping the benefits of infecting the host made vulnerable by the cooperators in the population. The authors hypothesize that an increase in frequency of cheaters may contribute to attenuation of virulence during serial propagation in the laboratory. The evolutionary dynamics of cheater frequency in a population have been explored in many contexts as part of kin selection theory. Cheaters can increase in a population by outcompeting cooperators in a host if overall relatedness within the invading population is low. Conversely, frequency of altruism, or costly cooperation, increases in a population if relatedness is high, which is enhanced by low effective dispersal. However, a population that is too isolated can suffer from inbreeding effects, and competition will occur mainly among relatives, which decreases the fitness benefits of altruism. Shapiro-Ilan and Raymond [1] tested changes in virulence and reproductive output in a serially propagated entomopathogenic nematode, Heterorhabditis floridensis. They compared lines of high or low relatedness, manipulated via multiplicity of infection (MOI) rates (where a low dose of nematodes gives high relatedness and a high dose gives low relatedness); and under global or local competition, manipulated by pooling populations emerging from all or only two host cadavers per generation, respectively. As predicted, treatments of high relatedness (low MOI) and global competition had the greatest level of reproduction, while all lines of low relatedness (high MOI) evolved decreased reproduction and decreased virulence, which led to extinction. The key finding was that lines in the high relatedness (low MOI) and low (local) competition treatment exhibited the most stable virulence through the 12 generations tested. Thus, to minimize attenuation of virulence while maintaining fitness of recently isolated entomopathogenic nematodes, the authors recommend insect hosts be inoculated with low doses of nematodes from inocula pools from as few cadavers as possible. The application of evolutionary theory, with a clever experimental design, to an important problem in pest management makes this paper particularly noteworthy. Reference [1] Shapiro-Ilan D, Raymond B. 2016. Limiting opportunities for cheating stabilizes virulence in insect parasitic nematodes. Evolutionary Applications 9:462-470. doi: 10.1111/eva.12348 | Limiting opportunities for cheating stabilizes virulence in insect parasitic nematodes | Shapiro-Ilan D. and B. Raymond | Cooperative secretion of virulence factors by pathogens can lead to social conflict when cheating mutants exploit collective secretion, but do not contribute to it. If cheats outcompete cooperators within hosts, this can cause loss of virulence.... | | Adaptation, Behavior & Social Evolution, Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory, Experimental Evolution, Population Genetics / Genomics, Reproduction and Sex | Thomas Sappington | 2016-12-15 18:33:39 | ||
05 Oct 2017

Using Connectivity To Identify Climatic Drivers Of Local AdaptationA new approach to identifying drivers of local adaptationRecommended by Ruth Arabelle Hufbauer based on reviews by Ruth Arabelle Hufbauer and Thomas LenormandLocal adaptation, the higher fitness a population achieves in its local “home” environment relative to other environments is a crucial phase in the divergence of populations, and as such both generates and maintains diversity. Local adaptation is enhanced by selection and genetic variation in the relevant traits, and decreased by gene flow and genetic drift. Demonstrating local adaptation is laborious, and is typically done with a reciprocal transplant design [1], documenting repeated geographic clines [e.g. 2, 3] also provides strong evidence of local adaptation. Even when well documented, it is often unknown which aspects of the environment impose selection. Indeed, differences in environment between different sites that are measured during studies of local adaptation explain little of the variance in the degree of local adaptation [4]. This poses a problem to population management. Given climate change and habitat destruction, understanding the environmental drivers of local adaptation can be crucially important to conducting successful assisted migration or targeted gene flow. In this manuscript, Macdonald et al. [5] propose a means of identifying which aspects of the environment select for local adaptation without conducting a reciprocal transplant experiment. The idea is that the strength of relationships between traits and environmental variables that are due to plastic responses to the environment will not be influenced by gene flow, but the strength of trait-environment relationships that are due to local adaptation should decrease with gene flow. This then can be used to reduce the somewhat arbitrary list of environmental variables on which data are available down to a targeted list more likely to drive local adaptation in specific traits. To perform such an analysis requires three things: 1) measurements of traits of interest in a species across locations, 2) an estimate of gene flow between locations, which can be replaced with a biologically meaningful estimate of how well connected those locations are from the point of view of the study species, and 3) data on climate and other environmental variables from across a species’ range, many of which are available on line. Macdonald et al. [5] demonstrate their approach using a skink (Lampropholis coggeri). They collected morphological and physiological data on individuals from multiple populations. They estimated connectivity among those locations using information on habitat suitability and dispersal potential [6], and gleaned climatic data from available databases and the literature. They find that two physiological traits, the critical minimum and maximum temperatures, show the strongest signs of local adaptation, specifically local adaptation to annual mean precipitation, precipitation of the driest quarter, and minimum annual temperature. These are then aspects of skink phenotype and skink habitats that could be explored further, or could be used to provide background information if migration efforts, for example for genetic rescue [7] were initiated. The approach laid out has the potential to spark a novel genre of research on local adaptation. It its simplest form, knowing that local adaptation is eroded by gene flow, it is intuitive to consider that if connectivity reduces the strength of the relationship between an environmental variable and a trait, that the trait might be involved in local adaptation. The approach is less intuitive than that, however – it relies not connectivity per-se, but the interaction between connectivity and different environmental variables and how that interaction alters trait-environment relationships. The authors lay out a number of useful caveats and potential areas that could use further development. It will be interesting to see how the community of evolutionary biologists responds. References [1] Blanquart F, Kaltz O, Nuismer SL and Gandon S. 2013. A practical guide to measuring local adaptation. Ecology Letters, 16: 1195-1205. doi: 10.1111/ele.12150 [2] Huey RB, Gilchrist GW, Carlson ML, Berrigan D and Serra L. 2000. Rapid evolution of a geographic cline in size in an introduced fly. Science, 287: 308-309. doi: 10.1126/science.287.5451.308 [3] Milesi P, Lenormand T, Lagneau C, Weill M and Labbé P. 2016. Relating fitness to long-term environmental variations in natura. Molecular Ecology, 25: 5483-5499. doi: 10.1111/mec.13855 [4] Hereford, J. 2009. A quantitative survey of local adaptation and fitness trade-offs. The American Naturalist 173: 579-588. doi: 10.1086/597611 [5] Macdonald SL, Llewelyn J and Phillips BL. 2017. Using connectivity to identify climatic drivers of local adaptation. bioRxiv, ver. 4 of October 4, 2017. doi: 10.1101/145169 [6] Macdonald SL, Llewelyn J, Moritz C and Phillips BL. 2017. Peripheral isolates as sources of adaptive diversity under climate change. Frontiers in Ecology and Evolution, 5:88. doi: 10.3389/fevo.2017.00088 [7] Whiteley AR, Fitzpatrick SW, Funk WC and Tallmon DA. 2015. Genetic rescue to the rescue. Trends in Ecology & Evolution, 30: 42-49. doi: 10.1016/j.tree.2014.10.009 | Using Connectivity To Identify Climatic Drivers Of Local Adaptation | Stewart L. Macdonald, John Llewelyn, Ben Phillips | Despite being able to conclusively demonstrate local adaptation, we are still often unable to objectively determine the climatic drivers of local adaptation. Given the rapid rate of global change, understanding the climatic drivers of local adapta... | | Adaptation, Evolutionary Applications | Ruth Arabelle Hufbauer | Thomas Lenormand | 2017-06-06 13:06:54 | |
12 Apr 2017

POSTPRINT
Genetic drift, purifying selection and vector genotype shape dengue virus intra-host genetic diversity in mosquitoesVectors as motors (of virus evolution)Recommended by Frédéric Fabre and Benoit MouryMany viruses are transmitted by biological vectors, i.e. organisms that transfer the virus from one host to another. Dengue virus (DENV) is one of them. Dengue is a mosquito-borne viral disease that has rapidly spread around the world since the 1940s. One recent estimate indicates 390 million dengue infections per year [1]. As many arthropod-borne vertebrate viruses, DENV has to cross several anatomical barriers in the vector, to multiply in its body and to invade its salivary glands before getting transmissible. As a consequence, vectors are not passive carriers but genuine hosts of the viruses that potentially have important effects on the composition of virus populations and, ultimately, on virus epidemiology and virulence. Within infected vectors, virus populations are expected to acquire new mutations and to undergo genetic drift and selection effects. However, the intensity of these evolutionary forces and the way they shape virus genetic diversity are poorly known. In their study, Lequime et al. [2] finely disentangled the effects of genetic drift and selection on DENV populations during their infectious cycle within mosquito (Aedes aegypti) vectors. They evidenced that the genetic diversity of viruses within their vectors is shaped by genetic drift, selection and vector genotype. The experimental design consisted in artificial acquisition of purified virus by mosquitoes during a blood meal. The authors monitored the diversity of DENV populations in Ae. aegypti individuals at different time points by high-throughput sequencing (HTS). They estimated the intensity of genetic drift and selection effects exerted on virus populations by comparing the DENV diversity at these sampling time points with the diversity in the purified virus stock (inoculum). Disentangling the effects of genetic drift and selection remains a methodological challenge because both evolutionary forces operate concomitantly and both reduce genetic diversity. However, selection reduces diversity in a reproducible manner among experimental replicates (here, mosquito individuals): the fittest variants are favoured at the expense of the weakest ones. In contrast, genetic drift reduces diversity in a stochastic manner among replicates. Genetic drift acts equally on all variants irrespectively of their fitness. The strength of genetic drift is frequently evaluated with the effective population size Ne: the lower Ne, the stronger the genetic drift [3]. The estimation of the effective population size of DENV populations by Lequime et al. [2] was based on single-nucleotide polymorphisms (SNPs) that were (i) present both in the inoculum and in the virus populations sampled at the different time points and (ii) that were neutral (or nearly-neutral) and therefore subjected to genetic drift only and insensitive to selection. As expected for viruses that possess small and constrained genomes, such neutral SNPs are extremely rare. Starting from a set of >1800 SNPs across the DENV genome, only three SNPs complied with the neutrality criteria and were enough represented in the sequence dataset for a precise Ne estimation. Using the method described by Monsion et al. [4], Lequime et al. [2] estimated Ne values ranging from 5 to 42 viral genomes (95% confidence intervals ranged from 2 to 161 founding viral genomes). Consequently, narrow bottlenecks occurred at the virus acquisition step, since the blood meal had allowed the ingestion of ca. 3000 infectious virus particles, on average. Interestingly, bottleneck sizes did not differ between mosquito genotypes. Monsion et al.’s [4] formula provides only an approximation of Ne. A corrected formula has been recently published [5]. We applied this exact Ne formula to the means and variances of the frequencies of the three neutral markers estimated before and after the bottlenecks (Table 1 in [2]), and nearly identical Ne estimates were obtained with both formulas. Selection intensity was estimated from the dN/dS ratio between the nonsynonymous and synonymous substitution rates using the HTS data on DENV populations. DENV genetic diversity increased following initial infection but was restricted by strong purifying selection during virus expansion in the midgut. Again, no differences were detected between mosquito genotypes. However and importantly, significant differences in DENV genetic diversity were detected among mosquito genotypes. As they could not be related to differences in initial genetic drift or to selection intensity, the authors raise interesting alternative hypotheses, including varying rates of de novo mutations due to differences in replicase fidelity or differences in the balancing selection regime. Interestingly, they also suggest that this observation could simply result from a methodological issue linked to the detection threshold of low-frequency SNPs. References [1] Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, et al. 2013. The global distribution and burden of dengue. Nature 496: 504–7 doi: 10.1038/nature12060 [2] Lequime S, Fontaine A, Gouilh MA, Moltini-Conclois I and Lambrechts L. 2016. Genetic drift, purifying selection and vector genotype shape dengue virus intra-host genetic diversity in mosquitoes. PloS Genetics 12: e1006111 doi: 10.1371/journal.pgen.1006111 [3] Charlesworth B. 2009. Effective population size and patterns of molecular evolution and variation. Nature Reviews Genetics 10: 195-205 doi: 10.1038/nrg2526 [4] Monsion B, Froissart R, Michalakis Y and Blanc S. 2008. Large bottleneck size in cauliflower mosaic virus populations during host plant colonization. PloS Pathogens 4: e1000174 doi: 10.1371/journal.ppat.1000174 [5] Thébaud G and Michalakis Y. 2016. Comment on ‘Large bottleneck size in cauliflower mosaic virus populations during host plant colonization’ by Monsion et al. (2008). PloS Pathogens 12: e1005512 doi: 10.1371/journal.ppat.1005512 | Genetic drift, purifying selection and vector genotype shape dengue virus intra-host genetic diversity in mosquitoes | Lequime S, Fontaine A, Gouilh MA, Moltini-Conclois I and Lambrechts L | Due to their error-prone replication, RNA viruses typically exist as a diverse population of closely related genomes, which is considered critical for their fitness and adaptive potential. Intra-host demographic fluctuations that stochastically re... | | Evolutionary Dynamics, Molecular Evolution, Population Genetics / Genomics | Frédéric Fabre | 2017-04-10 14:26:04 | ||
24 Oct 2019
Testing host-plant driven speciation in phytophagous insects : a phylogenetic perspectivePhylogenetic approaches for reconstructing macroevolutionary scenarios of phytophagous insect diversificationRecommended by Hervé Sauquet based on reviews by Brian O'Meara and 1 anonymous reviewerPlant-animal interactions have long been identified as a major driving force in evolution. However, only in the last two decades have rigorous macroevolutionary studies of the topic been made possible, thanks to the increasing availability of densely sampled molecular phylogenies and the substantial development of comparative methods. In this extensive and thoughtful perspective [1], Jousselin and Elias thoroughly review current hypotheses, data, and available macroevolutionary methods to understand how plant-insect interactions may have shaped the diversification of phytophagous insects. First, the authors review three main hypotheses that have been proposed to lead to host-plant driven speciation in phytophagous insects: the ‘escape and radiate’, ‘oscillation’, and ‘musical chairs’ scenarios, each with their own set of predictions. Jousselin and Elias then synthesize a vast core of recent studies on different clades of insects, where explicit phylogenetic approaches have been used. In doing so, they highlight heterogeneity in both the methods being used and predictions being tested across these studies and warn against the risk of subjective interpretation of the results. Lastly, they advocate for standardization of phylogenetic approaches and propose a series of simple tests for the predictions of host-driven speciation scenarios, including the characterization of host-plant range history and host breadth history, and diversification rate analyses. This helpful review will likely become a new point of reference in the field and undoubtedly help many researchers formalize and frame questions of plant-insect diversification in future studies of phytophagous insects. References [1] Jousselin, E., Elias, M. (2019). Testing Host-Plant Driven Speciation in Phytophagous Insects: A Phylogenetic Perspective. arXiv, 1910.09510, ver. 1 peer-reviewed and recommended by PCI Evol Biol. https://arxiv.org/abs/1910.09510v1 | Testing host-plant driven speciation in phytophagous insects : a phylogenetic perspective | Emmanuelle Jousselin, Marianne Elias | During the last two decades, ecological speciation has been a major research theme in evolutionary biology. Ecological speciation occurs when reproductive isolation between populations evolves as a result of niche differentiation. Phytophagous ins... | | Macroevolution, Phylogenetics / Phylogenomics, Speciation, Species interactions | Hervé Sauquet | 2019-02-25 17:31:33 | ||
31 Jul 2017

Selection on morphological traits and fluctuating asymmetry by a fungal parasite in the yellow dung flyParasite-mediated selection promotes small body size in yellow dung fliesRecommended by Rodrigo Medel based on reviews by Rodrigo Medel and 1 anonymous reviewerBody size has long been considered as one of the most important organismic traits influencing demographical processes, population size, and evolution of life history strategies [1, 2]. While many studies have reported a selective advantage of large body size, the forces that determine small-sized organisms are less known, and reports of negative selection coefficients on body size are almost absent at present. This lack of knowledge is unfortunate as climate change and energy demands in stressful environments, among other factors, may produce new selection scenarios and unexpected selection surfaces [3]. In this manuscript, Blanckenhorn [4] reports on a potential explanation for the surprising 10% body size decrease observed in a Swiss population of yellow dung flies during 1993 - 2009. The author took advantage of a fungus outbreak in 2002 to assess the putative role of the fungus Entomopthora scatophagae, a specific parasite of adult yellow dung flies, as selective force acting upon host body size. His findings indicate that, as expected by sexual selection theory, large males experience a mating advantage. However, this positive sexual selection is opposed by a strong negative selection on male and female body size through the viability fitness component. This study provides the first evidence of parasite-mediated disadvantage of large adult body size in the field. While further experimental work is needed to elucidate the exact causes of body size reduction in the population, the author proposes a variation of the trade-off hypothesis raised by Rantala & Roff [5] that large-sized individuals face an immunity cost due to their high absolute energy demands in stressful environments. References [1] Peters RH. 1983. The ecological implications of body size. Cambridge University Press, Cambridge. [2] Schmidt-Nielsen K. 1984. Scaling: why is animal size so important? Cambridge University Press, Cambridge. [3] Ohlberger J. 2013. Climate warming and ectotherm body size: from individual physiology to community ecology. Functional Ecology 27: 991-1001. doi: 10.1111/1365-2435.12098 [4] Blanckenhorn WU. 2017. Selection on morphological traits and fluctuating asymmetry by a fungal parasite in the yellow dung fly. bioRxiv 136325, ver. 2 of 29th June 2017. doi: 10.1101/136325 [5] Rantala MJ & Roff DA. 2005. An analysis of trade-offs in immune function, body size and development time in the Mediterranean field cricket, Gryllus bimaculatus. Functional Ecology 19: 323-330. doi: 10.1111/j.1365-2435.2005.00979.x | Selection on morphological traits and fluctuating asymmetry by a fungal parasite in the yellow dung fly | Wolf U. Blanckenhorn | Evidence for selective disadvantages of large body size remains scarce in general. Previous phenomenological studies of the yellow dung fly *Scathophaga stercoraria* have demonstrated strong positive sexual and fecundity selection on male and fema... | | Behavior & Social Evolution, Evolutionary Ecology, Life History, Sexual Selection | Rodrigo Medel | Rodrigo Medel | 2017-05-10 11:16:26 | |
03 Aug 2017
POSTPRINT
Fisher's geometrical model and the mutational patterns of antibiotic resistance across dose gradientsWhat doesn’t kill us makes us stronger: can Fisher’s Geometric model predict antibiotic resistance evolution?Recommended by Inês Fragata and Claudia Bank
The increasing number of reported cases of antibiotic resistance is one of today’s major public health concerns. Dealing with this threat involves understanding what drives the evolution of antibiotic resistance and investigating whether we can predict (and subsequently avoid or circumvent) it [1]. References [1] Palmer AC, and Kishony R. 2013. Understanding, predicting and manipulating the genotypic evolution of antibiotic resistance. Nature Review Genetics 14: 243—248. doi: 10.1038/nrg3351 [2] Tenaillon O. 2014. The utility of Fisher’s geometric model in evolutionary genetics. Annual Review of Ecology, Evolution and Systematics 45: 179—201. doi: 10.1146/annurev-ecolsys-120213-091846 [3] Blanquart F and Bataillon T. 2016. Epistasis and the structure of fitness landscapes: are experimental fitness landscapes compatible with Fisher’s geometric model? Genetics 203: 847—862. doi: 10.1534/genetics.115.182691 [4] Harmand N, Gallet R, Jabbour-Zahab R, Martin G and Lenormand T. 2017. Fisher’s geometrical model and the mutational patterns of antibiotic resistance across dose gradients. Evolution 71: 23—37. doi: 10.1111/evo.13111 [5] de Visser, JAGM, and Krug J. 2014. Empirical fitness landscapes and the predictability of evolution. Nature 15: 480—490. doi: 10.1038/nrg3744 [6] Palmer AC, Toprak E, Baym M, Kim S, Veres A, Bershtein S and Kishony R. 2015. Delayed commitment to evolutionary fate in antibiotic resistance fitness landscapes. Nature Communications 6: 1—8. doi: 10.1038/ncomms8385 | Fisher's geometrical model and the mutational patterns of antibiotic resistance across dose gradients | Noémie Harmand, Romain Gallet, Roula Jabbour-Zahab, Guillaume Martin, Thomas Lenormand | Fisher's geometrical model (FGM) has been widely used to depict the fitness effects of mutations. It is a general model with few underlying assumptions that gives a large and comprehensive view of adaptive processes. It is thus attractive in sever... | | Adaptation | Inês Fragata | 2017-08-01 16:06:02 | ||
15 Dec 2016

POSTPRINT
Basidiomycete yeasts in the cortex of ascomycete macrolichensNew partner at the core of macrolichen diversityRecommended by Enric Frago and Benoit FaconIt has long been known that most multicellular eukaryotes rely on microbial partners for a variety of functions including nutrition, immune reactions and defence against enemies. Lichens are probably the most popular example of a symbiosis involving a photosynthetic microorganism (an algae, a cyanobacteria or both) living embedded within the filaments of a fungus (usually an ascomycete). The latter is the backbone structure of the lichen, whereas the former provides photosynthetic products. Lichens are unique among symbioses because the structures the fungus and the photosynthetic microorganism form together do not resemble any of the two species living in isolation. Classic textbook examples like lichens are not often challenged and this is what Toby Spribille and his co-authors did with their paper published in July 2016 in Science [1]. This story started with the study of two species of macrolichens from the class of Lecanoromycetes that are commonly found in the mountains of Montana (US): Bryoria fremontii and B. tortuosa. For more than 90 years, these species have been known to differ in their chemical composition and colour, but studies performed so far failed in finding differences at the molecular level in both the mycobiont and the photobiont. These two species were therefore considered as nomenclatural synonyms, and the origin of their differences remained elusive. To solve this mystery, the authors of this work performed a transcriptome-wide analysis that, relative to previous studies, expanded the taxonomic range to all Fungi. This analysis revealed higher abundances of a previously unknown basidiomycete yeast from the genus Cyphobasidium in one of the lichen species, a pattern that was further confirmed by combining microscopy imaging and the fluorescent in situ hybridisation technique (FISH). Finding out that a previously unknown micro-organism changes the colour and the chemical composition of an organism is surprising but not new. For instance, bacterial symbionts are able to trigger colour changes in some insect species [2], and endophyte fungi are responsible for the production of defensive compounds in the leaves of several grasses [3]. The study by Spribille and his co-authors is fascinating because it demonstrates that Cyphobasidium yeasts have played a key role in the evolution and diversification of Lecanoromycetes, one of the most diverse classes of macrolichens. Indeed these basidiomycete yeasts were not only found in Bryoria but in 52 other lichen genera from all six continents, and these included 42 out of 56 genera in the family Parmeliaceae. Most of these sequences formed a highly supported monophyletic group, and a molecular clock revealed that the origin of many macrolichen groups occurred around the same time Cyphobasidium yeasts split from Cystobasidium, their nearest relatives. This newly discovered passenger is therefore an ancient inhabitant of lichens and has driven the evolution of this emblematic group of organisms. This study raises an important question on the stability of complex symbiotic partnerships. In intimate obligatory symbioses the evolutionary interests of both partners are often identical and what is good for one is also good for the other. This is the case of several insects that feed on poor diets like phloem and xylem sap, and which carry vertically-transmitted symbionts that provide essential nutrients. Molecular phylogenetic studies have repeatedly shown that in several insect groups transition to phloem or xylem feeding occurred at the same time these nutritional symbionts were acquired [4]. In lichens, an outstanding question is to know what was the key feature Cyphobasidium yeasts brought to the symbiosis. As suggested by the authors, these yeasts are likely to be involved in the production of secondary defensive metabolites and architectural structures, but, are these services enough to explain the diversity found in macrolichens? This paper is an appealing example of a multipartite symbiosis where the different partners share an ancient evolutionary history. References [1] Spribille T, Tuovinen V, Resl P, et al. 2016. Basidiomycete yeasts in the cortex of ascomycete macrolichens. Science 353:488–92. doi: 10.1126/science.aaf8287 [2] Tsuchida T, Koga R, Horikawa M, et al. 2010. Symbiotic Bacterium Modifies Aphid Body Color. Science 330:1102–1104. doi: 10.1126/science.1195463 [3] Clay K. 1988. Fungal Endophytes of Grasses: A Defensive Mutualism between Plants and Fungi. Ecology 69:10–16. doi: 10.2307/1943155 [4] Moran NA. 2007. Symbiosis as an adaptive process and source of phenotypic complexity. Proceeding of the National Academy of Science USA 104:8627–8633. doi: 10.1073/pnas.0611659104 | Basidiomycete yeasts in the cortex of ascomycete macrolichens | Spribille T, Tuovinen V, Resl P, et al. | For over 140 years, lichens have been regarded as a symbiosis between a single fungus, usually an ascomycete, and a photosynthesizing partner. Other fungi have long been known to occur as occasional parasites or endophytes, but the one lichen–one ... | | Adaptation, Evolutionary Ecology, Genome Evolution, Genotype-Phenotype, Life History, Macroevolution, Molecular Evolution, Phylogenetics / Phylogenomics, Speciation, Species interactions | Enric Frago | 2016-12-15 05:46:14 |




