
Latest recommendations

| Id | Title * | Authors * | Abstract * | Picture * ▲ | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
18 Jan 2023
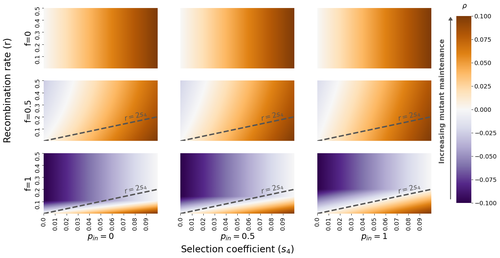
The fate of recessive deleterious or overdominant mutations near mating-type loci under partial selfingMaintenance of deleterious mutations and recombination suppression near mating-type loci under selfingRecommended by Aurelien Tellier based on reviews by 3 anonymous reviewers based on reviews by 3 anonymous reviewers
The causes and consequences of the evolution of sexual reproduction are a major topic in evolutionary biology. With advances in sequencing technology, it becomes possible to compare sexual chromosomes across species and infer the neutral and selective processes shaping polymorphism at these chromosomes. Most sex and mating-type chromosomes exhibit an absence of recombination in large genomic regions around the animal, plant or fungal sex-determining genes. This suppression of recombination likely occurred in several time steps generating stepwise increasing genomic regions starting around the sex-determining genes. This mechanism generates so-called evolutionary strata of differentiation between sex chromosomes (Nicolas et al., 2004, Bergero and Charlesworth, 2009, Hartmann et al. 2021). The evolution of extended regions of recombination suppression is also documented on mating-type chromosomes in fungi (Hartmann et al., 2021) and around supergenes (Yan et al., 2020, Jay et al., 2021). The exact reason and evolutionary mechanisms for this phenomenon are still, however, debated. Two hypotheses are proposed: 1) sexual antagonism (Charlesworth et al., 2005), which, nevertheless, does explain the observed occurrence of the evolutionary strata, and 2) the sheltering of deleterious alleles by inversions carrying a lower load than average in the population (Charlesworth and Wall, 1999, Antonovics and Abrams, 2004). In the latter, the mechanism is as follows. A genetic inversion or a suppressor of recombination in cis may exhibit some overdominance behaviour. The inversion exhibiting less recessive deleterious mutations (compared to others at the same locus) may increase in frequency, before at higher frequency occurring at the homozygous state, expressing its genetic load. However, the inversion may never be at the homozygous state if it is genetically linked to a gene in a permanently heterozygous state. The inversion can then be advantageous and may reach fixation at the sex chromosome (Charlesworth and Wall, 1999, Antonovics and Abrams, 2004, Jay et al., 2022). These selective mechanisms promote thus the suppression of recombination around the sex-determining gene, and recessive deleterious mutations are permanently sheltered. This hypothesis is corroborated by the sheltering of deleterious mutations observed around loci under balancing selection (Llaurens et al. 2009, Lenz et al. 2016) and around mating-type genes in fungi and supergenes (Jay et al. 2021, Jay et al., 2022). In this present theoretical study, Tezenas et al. (2022) analyse how linkage to a necessarily heterozygous fungal mating type locus influences the persistence/extinction time of a new mutation at a second selected locus. This mutation can either be deleterious and recessive, or overdominant. There is arbitrary linkage between the two loci, and sexual reproduction occurs either between 1) gametes of different individuals (outcrossing), or 2) by selfing with gametes originating from the same (intra-tetrad) or different (inter-tetrad) tetrads produced by that individual. Note, here, that the mating-type gene does not prevent selfing. The authors study the initial stochastic dynamics of the mutation using a multi-type branching process (and simulations when analytical results cannot be obtained) to compute the extinction time of the deleterious mutation. The main result is that the presence of a mating-type locus always decreases the purging probability and increases the purging time of the mutations under selfing. Ultimately, deleterious mutations can indeed accumulate near the mating-type locus over evolutionary time scales. In a nutshell, high selfing or high intra-tetrad mating do increase the sheltering effect of the mating-type locus. In effect, the outcome of sheltering of deleterious mutations depends on two opposing mechanisms: 1) a higher selfing rate induces a greater production of homozygotes and an increased effect of the purging of deleterious mutations, while 2) a higher intra-tetrad selfing rate (or linkage with the mating-type locus) generates heterozygotes which have a small genetic load (and are favoured). The authors also show that rare events of extremely long maintenance of deleterious mutations can occur. The authors conclude by highlighting the manifold effect of selfing which reduces the effective population size and thus impairs the efficiency of selection and increases the mutational load, while also favouring the purge of deleterious homozygous mutations. Furthermore, this study emphasizes the importance of studying the maintenance and accumulation of deleterious mutations in the vicinity of heterozygous loci (e.g. under balancing selection) in selfing species. References Antonovics J, Abrams JY (2004) Intratetrad Mating and the Evolution of Linkage Relationships. Evolution, 58, 702–709. https://doi.org/10.1111/j.0014-3820.2004.tb00403.x Bergero R, Charlesworth D (2009) The evolution of restricted recombination in sex chromosomes. Trends in Ecology & Evolution, 24, 94–102. https://doi.org/10.1016/j.tree.2008.09.010 Charlesworth D, Morgan MT, Charlesworth B (1990) Inbreeding Depression, Genetic Load, and the Evolution of Outcrossing Rates in a Multilocus System with No Linkage. Evolution, 44, 1469–1489. https://doi.org/10.1111/j.1558-5646.1990.tb03839.x Charlesworth D, Charlesworth B, Marais G (2005) Steps in the evolution of heteromorphic sex chromosomes. Heredity, 95, 118–128. https://doi.org/10.1038/sj.hdy.6800697 Charlesworth B, Wall JD (1999) Inbreeding, heterozygote advantage and the evolution of neo–X and neo–Y sex chromosomes. Proceedings of the Royal Society of London. Series B: Biological Sciences, 266, 51–56. https://doi.org/10.1098/rspb.1999.0603 Hartmann FE, Duhamel M, Carpentier F, Hood ME, Foulongne-Oriol M, Silar P, Malagnac F, Grognet P, Giraud T (2021) Recombination suppression and evolutionary strata around mating-type loci in fungi: documenting patterns and understanding evolutionary and mechanistic causes. New Phytologist, 229, 2470–2491. https://doi.org/10.1111/nph.17039 Jay P, Chouteau M, Whibley A, Bastide H, Parrinello H, Llaurens V, Joron M (2021) Mutation load at a mimicry supergene sheds new light on the evolution of inversion polymorphisms. Nature Genetics, 53, 288–293. https://doi.org/10.1038/s41588-020-00771-1 Jay P, Tezenas E, Véber A, Giraud T (2022) Sheltering of deleterious mutations explains the stepwise extension of recombination suppression on sex chromosomes and other supergenes. PLOS Biology, 20, e3001698. https://doi.org/10.1371/journal.pbio.3001698 Lenz TL, Spirin V, Jordan DM, Sunyaev SR (2016) Excess of Deleterious Mutations around HLA Genes Reveals Evolutionary Cost of Balancing Selection. Molecular Biology and Evolution, 33, 2555–2564. https://doi.org/10.1093/molbev/msw127 Llaurens V, Gonthier L, Billiard S (2009) The Sheltered Genetic Load Linked to the S Locus in Plants: New Insights From Theoretical and Empirical Approaches in Sporophytic Self-Incompatibility. Genetics, 183, 1105–1118. https://doi.org/10.1534/genetics.109.102707 Nicolas M, Marais G, Hykelova V, Janousek B, Laporte V, Vyskot B, Mouchiroud D, Negrutiu I, Charlesworth D, Monéger F (2004) A Gradual Process of Recombination Restriction in the Evolutionary History of the Sex Chromosomes in Dioecious Plants. PLOS Biology, 3, e4. https://doi.org/10.1371/journal.pbio.0030004 Tezenas E, Giraud T, Véber A, Billiard S (2022) The fate of recessive deleterious or overdominant mutations near mating-type loci under partial selfing. bioRxiv, 2022.10.07.511119, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.10.07.511119 Yan Z, Martin SH, Gotzek D, Arsenault SV, Duchen P, Helleu Q, Riba-Grognuz O, Hunt BG, Salamin N, Shoemaker D, Ross KG, Keller L (2020) Evolution of a supergene that regulates a trans-species social polymorphism. Nature Ecology & Evolution, 4, 240–249. https://doi.org/10.1038/s41559-019-1081-1 | The fate of recessive deleterious or overdominant mutations near mating-type loci under partial selfing | Emilie Tezenas, Tatiana Giraud, Amandine Veber, Sylvain Billiard | <p style="text-align: justify;">Large regions of suppressed recombination having extended over time occur in many organisms around genes involved in mating compatibility (sex-determining or mating-type genes). The sheltering of deleterious alleles... | | Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Theory, Genome Evolution, Population Genetics / Genomics, Reproduction and Sex | Aurelien Tellier | 2022-10-10 13:50:30 | ||
21 Nov 2018
Convergent evolution as an indicator for selection during acute HIV-1 infectionIs convergence an evidence for positive selection?Recommended by Guillaume Achaz based on reviews by Jeffrey Townsend and 1 anonymous reviewerThe preprint by Bertels et al. [1] reports an interesting application of the well-accepted idea that positively selected traits (here variants) can appear several times independently; think about the textbook examples of flight capacity. Hence, the authors assume that reciprocally convergence implies positive selection. The methodology becomes then, in principle, straightforward as one can simply count variants in independent datasets to detect convergent mutations. References [1] Bertels, F., Metzner, K. J., & Regoes R. R. (2018). Convergent evolution as an indicator for selection during acute HIV-1 infection. BioRxiv, 168260, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/168260 | Convergent evolution as an indicator for selection during acute HIV-1 infection | Frederic Bertels, Karin J Metzner, Roland R Regoes | <p>Convergent evolution describes the process of different populations acquiring similar phenotypes or genotypes. Complex organisms with large genomes only rarely and only under very strong selection converge to the same genotype. In contrast, ind... | | Bioinformatics & Computational Biology, Evolutionary Applications, Genome Evolution, Molecular Evolution | Guillaume Achaz | 2017-07-26 08:39:17 | ||
16 Dec 2022
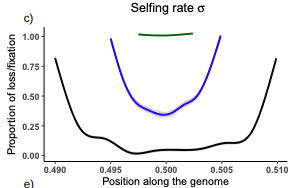
Conditions for maintaining and eroding pseudo-overdominance and its contribution to inbreeding depressionPseudo-overdominance: how linkage and selection can interact and oppose to purging of deleterious mutations.Recommended by Sylvain Glémin based on reviews by Yaniv Brandvain, Lei Zhao and 1 anonymous reviewerMost mutations affecting fitness are deleterious and they have many evolutionary consequences. The dynamics and consequences of deleterious mutations are a long-standing question in evolutionary biology and a strong theoretical background has already been developed, for example, to predict the mutation load, inbreeding depression or background selection. One of the classical results is that inbreeding helps purge partially recessive deleterious mutations by exposing them to selection in homozygotes. However, this mainly results from single-locus considerations. When interactions among several, more or less linked, deleterious mutations are taken into account, peculiar dynamics can emerge. One of them, called pseudo-overdominance (POD), corresponds to the maintenance in a population of two (or more) haplotype blocks composed of several recessive deleterious mutations in repulsion that mimics overdominance. Indeed, homozygote individuals for one of the haplotype blocks expose many deleterious mutations to selection whereas they are reciprocally masked in heterozygotes, leading to higher fitness of heterozygotes compared to both homozygotes. A related process, called associative overdominance (AOD) is the effect of such deleterious alleles in repulsion on the linked neutral variation that can be increased by AOD. Although this possibility has been recognized for a long time (Otha and Kimura 1969), it has been mainly considered an anecdotal process. Recently, both theoretical (Zhao and Charlesworth 2016) and genomic analyses (Gilbert et al. 2020) have renewed interest in such a process, suggesting that it could be important in weakly recombining regions of a genome. Donald Waller (2021) - one of the co-authors of the current work - also recently proposed that POD could be quantitatively important with broad implications, and could resolve some unexplained observations such as the maintenance of inbreeding depression in highly selfing species. Yet, a proper theoretical framework analysing the effect of inbreeding on POD was lacking. In this theoretical work, Diala Abu Awad and Donald Waller (2022) addressed this question through an elegant combination of analytical predictions and intensive multilocus simulations. They determined the conditions under which POD can be maintained and how long it could resist erosion by recombination, which removes the negative association between deleterious alleles (repulsion) at the core of the mechanism. They showed that under tight linkage, POD regions can persist for a long time and generate substantial segregating load and inbreeding depression, even under inbreeding, so opposing (for a while) to the purging effect. They also showed that background selection can affect the genomic structure of POD regions by rapidly erasing weak POD regions but maintaining strong POD regions (i.e with many tightly linked deleterious alleles). These results have several implications. They can explain the maintenance of inbreeding depression despite inbreeding (as anticipated by Waller 2021), which has implications for the evolution of mating systems. If POD can hardly emerge under high selfing, it can persist from an outcrossing ancestor long after the transition towards a higher selfing rate and could explain the maintenance of mixed mating systems(which is possible with true overdominance, see Uyenoyama and Waller 1991). The results also have implications for genomic analyses, pointing to regions of low or no recombination where POD could be maintained, generating both higher diversity and heterozygosity than expected and variance in fitness. As structural variations are likely widespread in genomes with possible effects on suppressing recombination (Mérot et al. 2020), POD regions should be checked more carefully in genomic analyses (see also Gilbert et al. 2020). Overall, this work should stimulate new theoretical and empirical studies, especially to assess how quantitatively strong and widespread POD can be. It also stresses the importance of properly considering genetic linkage genome-wide, and so the role of recombination landscapes in determining patterns of diversity and fitness effects. References
Awad DA, Waller D (2022) Conditions for maintaining and eroding pseudo-overdominance and its contribution to inbreeding depression. bioRxiv, 2021.12.16.473022, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.12.16.473022 Gilbert KJ, Pouyet F, Excoffier L, Peischl S (2020) Transition from Background Selection to Associative Overdominance Promotes Diversity in Regions of Low Recombination. Current Biology, 30, 101-107.e3. https://doi.org/10.1016/j.cub.2019.11.063 Mérot C, Oomen RA, Tigano A, Wellenreuther M (2020) A Roadmap for Understanding the Evolutionary Significance of Structural Genomic Variation. Trends in Ecology & Evolution, 35, 561–572. https://doi.org/10.1016/j.tree.2020.03.002 Ohta T, Kimura M (1969) Linkage disequilibrium at steady state determined by random genetic drift and recurrent mutation. Genetics, 63, 229–238. https://doi.org/10.1093/genetics/63.1.229 Uyenoyama MK, Waller DM (1991) Coevolution of self-fertilization and inbreeding depression II. Symmetric overdominance in viability. Theoretical Population Biology, 40, 47–77. https://doi.org/10.1016/0040-5809(91)90046-I Waller DM (2021) Addressing Darwin’s dilemma: Can pseudo-overdominance explain persistent inbreeding depression and load? Evolution, 75, 779–793. https://doi.org/10.1111/evo.14189 Zhao L, Charlesworth B (2016) Resolving the Conflict Between Associative Overdominance and Background Selection. Genetics, 203, 1315–1334. https://doi.org/10.1534/genetics.116.188912 | Conditions for maintaining and eroding pseudo-overdominance and its contribution to inbreeding depression | Diala Abu Awad, Donald Waller | <p style="text-align: justify;">Classical models that ignore linkage predict that deleterious recessive mutations should purge or fix within inbred populations, yet inbred populations often retain moderate to high segregating load. True overdomina... | | Evolutionary Dynamics, Evolutionary Theory, Genome Evolution, Hybridization / Introgression, Population Genetics / Genomics, Reproduction and Sex | Sylvain Glémin | 2022-01-04 12:15:35 | ||
30 May 2023
slendr: a framework for spatio-temporal population genomic simulations on geographic landscapesA new powerful tool to easily encode the geo-spatial dimension in population genetics simulationsRecommended by Emiliano Trucchi based on reviews by Liisa Loog and 2 anonymous reviewers
Models explaining the evolutionary processes operating in living beings are often impossible to test in the real world. This is mainly because of the long time (i.e., the number of generations) which is necessary for evolution to unfold. In addition, any such experiment would require a large number of individuals and, more importantly, many replicates to account for the inherent variance of the evolutionary processes under investigation. Only organisms with fast generation times and favourable rearing conditions can be used to explicitly test for specific evolutionary hypotheses. Computer simulations have filled this gap, revolutionising experimental testing in evolutionary biology by integrating genetic models into complex population dynamics, which can be run for (potentially) any length of time. Without going into an extensive description of the many available approaches for population genetics simulations (an exhaustive review can be found in Hoban et al 2012), three main aspects are, in my opinion, important for categorising and choosing one simulation approach over another. The first concerns the basic distinction between coalescent-based and individual-based simulators: the former being an efficient approach, which simulates back in time the coalescence events of a sample of homologous DNA fragments, while the latter is a more computationally intensive approach where all of the individuals (and their underlying genetic/genomic features) in the population are simulated forward-in-time, generation after generation. The second aspect concerns the simulation of natural selection. Although natural selection can be integrated into backward-in-time simulations, it is more realistically implemented as individual-based fitness in forward-in-time simulators. The third point, which has been often overlooked in evolutionary simulations, is about the possibility to design a simulation scenario where individuals and populations can exploit a physical (geographical) space. Amongst the coalescent-based simulators, SPLATCHE (Currat et al 2004), and its derivatives, is one of the few simulation tools deploying the coalescence process in sub-demes which are all connected by migration, thus getting as close as possible to a spatially-explicit population. On the other hand, individual-based simulators, whose development followed the increasing power of computational machines, offer a great opportunity to include spatio-temporal dynamics within a genomic simulation model. One of the most realistic and efficient individual-based forward-in-time simulators available is SLiM (Haller and Messer 2017), which allows users to implement simulations in arbitrarily complex spaces. Here, the more challenging part is encoding the spatially-explicit scenarios using the SLiM-specific EIDOS language. The new R package slendr (Petr et al 2022) offers a practical solution to this issue. By wrapping different tools into a well-known scripting language, slendr allows the design of spatiotemporal simulation scenarios which can be directly executed in the individual-based SLiM simulator, and the output stored with modern tree-sequence analysis tools (tskit; Kellerer et al 2018). Alternatively, simulations of non-spatial models can be run using a coalescent-based algorithm (msprime; Baumdicker et al 2022). The main advantage of slendr is that the whole simulative experiment can be performed entirely in the R environment, taking advantage of the many libraries available for geospatial and genomic data analysis, statistics, and visualisation. The open-source nature of this package, whose main aim is to make complex population genomics modelling more accessible, and the vibrant community of SLiM and tskit users will very likely make slendr widely used amongst the molecular ecology and evolutionary biology communities. Slendr handles real Earth cartographic data where users can design realistic demographic processes which characterise natural populations (i.e., expansions, displacement of large populations, interactions among populations, migrations, population splits, etc.) by changing spatial population boundaries across time and space. All in all, slendr is a very flexible and scalable framework to test the accuracy of spatial models, hypotheses about demography and selection, and interactions between organisms across space and time. REFERENCES Baumdicker, F., Bisschop, G., Goldstein, D., Gower, G., Ragsdale, A. P., Tsambos, G., ... & Kelleher, J. (2022). Efficient ancestry and mutation simulation with msprime 1.0. Genetics, 220(3), iyab229. https://doi.org/10.1093/genetics/iyab229 Currat, M., Ray, N., & Excoffier, L. (2004). SPLATCHE: a program to simulate genetic diversity taking into account environmental heterogeneity. Molecular Ecology Notes, 4(1), 139-142. https://doi.org/10.1046/j.1471-8286.2003.00582.x Haller, B. C., & Messer, P. W. (2017). SLiM 2: flexible, interactive forward genetic simulations. Molecular biology and evolution, 34(1), 230-240. https://doi.org/10.1093/molbev/msw211 Hoban, S., Bertorelle, G., & Gaggiotti, O. E. (2012). Computer simulations: tools for population and evolutionary genetics. Nature Reviews Genetics, 13(2), 110-122. https://doi.org/10.1038/nrg3130 Kelleher, J., Thornton, K. R., Ashander, J., & Ralph, P. L. (2018). Efficient pedigree recording for fast population genetics simulation. PLoS computational biology, 14(11), e1006581. https://doi.org/10.1371/journal.pcbi.1006581 Petr, M., Haller, B. C., Ralph, P. L., & Racimo, F. (2023). slendr: a framework for spatio-temporal population genomic simulations on geographic landscapes. bioRxiv, 2022.03.20.485041, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.03.20.485041 | slendr: a framework for spatio-temporal population genomic simulations on geographic landscapes | Martin Petr, Benjamin C. Haller, Peter L. Ralph, Fernando Racimo | <p style="text-align: justify;">One of the goals of population genetics is to understand how evolutionary forces shape patterns of genetic variation over time. However, because populations evolve across both time and space, most evolutionary proce... | | Bioinformatics & Computational Biology, Evolutionary Theory, Phylogeography & Biogeography, Population Genetics / Genomics | Emiliano Trucchi | 2022-09-14 12:57:56 | ||
17 Feb 2020
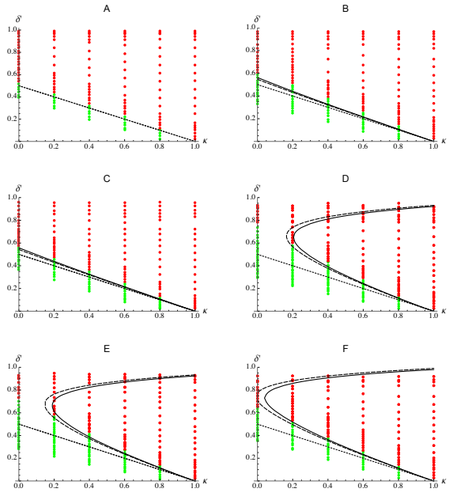
Epistasis, inbreeding depression and the evolution of self-fertilizationEpistasis and the evolution of selfingRecommended by Sylvain Gandon based on reviews by Nick Barton and 1 anonymous reviewerThe evolution of selfing results from a balance between multiple evolutionary forces. Selfing provides an "automatic advantage" due to the higher efficiency of selfers to transmit their genes via selfed and outcrossed offspring. Selfed offspring, however, may suffer from inbreeding depression. In principle the ultimate evolutionary outcome is easy to predict from the relative magnitude of these two evolutionary forces [1,2]. Yet, several studies explicitly taking into account the genetic architecture of inbreeding depression noted that these predictions are often too restrictive because selfing can evolve in a broader range of conditions [3,4]. References [1] Holsinger, K. E., Feldman, M. W., and Christiansen, F. B. (1984). The evolution of self-fertilization in plants: a population genetic model. The American Naturalist, 124(3), 446-453. doi: 10.1086/284287 | Epistasis, inbreeding depression and the evolution of self-fertilization | Diala Abu Awad and Denis Roze | <p>Inbreeding depression resulting from partially recessive deleterious alleles is thought to be the main genetic factor preventing self-fertilizing mutants from spreading in outcrossing hermaphroditic populations. However, deleterious alleles may... | | Evolutionary Theory, Quantitative Genetics, Reproduction and Sex | Sylvain Gandon | 2019-10-18 09:29:41 | ||
21 Nov 2022

Artisanal and farmers bread making practices differently shape fungal species community composition in French sourdoughsThe variety of bread-making practices promotes diversity conservation in food microbial communitiesRecommended by Tatiana Giraud and Jeanne Ropars based on reviews by 2 anonymous reviewers
Domesticated organisms are excellent models for understanding ecology and evolution and they are important for our food production and safety. While less studied than plants and animals, micro-organisms have also been domesticated, in particular for food fermentation [1]. The most studied domesticated micro-organism is the yeast used to make wine, beer and bread, Saccharomyces cerevisiae [2, 3, 4]. Filamentous fungi used for cheese-making have recently gained interest, for example Penicillium roqueforti used to make blue cheeses and P. camemberti to make soft cheeses [5, 6, 7, 8]. As for plants and animals, domestication has led to beneficial traits for food production in fermenting fungi, but also to bottlenecks and degeneration [6, 7, 9]; P. camemberti for example does not produce enough spores any more for optimal culture and inoculation and P. roqueforti has lost sexual fertility [9]. The loss of genetic diversity and of species diversity in our food production system is concerning for multiple reasons : i) it jeopardizes future improvement in the face of global changes ; ii) it causes the loss of evolved diversity during centuries under human selection, and therefore of beneficial characteristics and specificities that we may never be able to recover ; iii) it leads to degeneration in the few cultivated strains; iv) it impoverishes the diversity of our food products and local adaptation of production practices. The study of domesticated fungi used for food fermentation has focused so far on the evolution of lineages and on their metabolic specificities. Microbiological assemblages and species diversity have been much less studied, while they likely also have a strong impact on the quality and safety of final products. This study by Elisa Michel and colleagues [10] addresses this question, using an interdisciplinary participatory research approach including bakers, psycho-sociologists and microbiologists to analyse bread-making practices and their impact on microbial communities in sourdough. Elisa Michel and colleagues [10] identified two distinct groups of bread-making practices based on interviews and surveys, with farmer-like practices (low bread production, use of ancient wheat populations, manual kneading, working at ambient temperature, long fermentation periods and no use of commercial baker’s yeast) versus more intensive, artisanal-like practices. Metabarcoding and microbial culture-based analyses showed that the well-known baker’s yeast, Saccharomyces cerevisiae, was, surprisingly, not the most common species in French organic sourdoughs. Kazachstania was the most represented yeast genus over all sourdoughs, both in terms of read abundance and of species diversity. Kazachstania species were also often dominant in individual sourdoughs, but Saccharomyces uvarum or Torulaspora delbrueckii could also be the dominant yeast species. Metabarcoding analyses further revealed that the composition of the fungal communities differed between the farmer-like and more intensive practices, representing the first evidence of the influence of artisanal practices on microbial communities. The fungal communities were impacted by a combination of bread-making variables including the type of wheat varieties, the length of fermentation, the quantity of bread made per week and the use of commercial yeast. Maintaining on farm less intensive bread-making practices, may allow the preservation of typical species and phenotypic diversity in microbial communities in sourdough. Farmer-like practices did not lead to higher diversity within sourdoughs but, overall, the diversity of bread-making practices allow maintaining a larger diversity in sourdoughs. For example, different Kazachstania species were most abundant in sourdoughs from artisanal-like and farmer-like practices. Interviews with the bakers suggested the role of dispersal of Kazachstania species in shaping sourdough microbial communities, dispersal occurring by seed exchanges, sourdough mixing or gifts, bread-making training in common or working in one another’s bakery. Nikolai Vavilov [11] had already highlighted for crops the importance of isolated cultures and selection in different farms for generating and preserving crop diversity, but also the importance of seed exchange for fostering adaptation. Furthermore, one of the yeast frequently found in artisanal sourdoughs, Kazachstania humilis, displayed phenotypic differences between sourdough and non-sourdough strains, suggesting domestication. The sourdough strains exhibited significantly higher CO2 production rate and a lower fermentation latency-phase time. The study by Elisa Michel and colleagues [10] is thus novel and inspiring in showing the importance of interdisciplinary studies, combining metabarcoding, microbiology and interviews for assessing the composition and diversity of microbial communities in human-made food, and in revealing the impact of artisanal-like bread-making practices in preserving microbial community diversity. Interdisciplinary studies are still rare but have already shown the importance of combining ethno-ecology, biology and evolution to decipher the role of human practices on genetic diversity in crops, animals and food microorganisms and to help preserving genetic resources [12]. For example, in the case of the bread wheat Triticum aestivum, such interdisciplinary studies have shown that genetic diversity has been shaped by farmers’ seed diffusion and farming practices [13]. We need more of such interdisciplinary studies on the impact of farmer versus industrial agricultural and food-making practices as we urgently need to preserve the diversity of micro-organisms used in food production that we are losing at a rapid pace [6, 7, 14]. References [1] Dupont J, Dequin S, Giraud T, Le Tacon F, Marsit S, Ropars J, Richard F, Selosse M-A (2017) Fungi as a Source of Food. Microbiology Spectrum, 5, 5.3.09. https://doi.org/10.1128/microbiolspec.FUNK-0030-2016 [2] Legras J-L, Galeote V, Bigey F, Camarasa C, Marsit S, Nidelet T, Sanchez I, Couloux A, Guy J, Franco-Duarte R, Marcet-Houben M, Gabaldon T, Schuller D, Sampaio JP, Dequin S (2018) Adaptation of S. cerevisiae to Fermented Food Environments Reveals Remarkable Genome Plasticity and the Footprints of Domestication. Molecular Biology and Evolution, 35, 1712–1727. https://doi.org/10.1093/molbev/msy066 [3] Bai F-Y, Han D-Y, Duan S-F, Wang Q-M (2022) The Ecology and Evolution of the Baker’s Yeast Saccharomyces cerevisiae. Genes, 13, 230. https://doi.org/10.3390/genes13020230 [4] Fay JC, Benavides JA (2005) Evidence for Domesticated and Wild Populations of Saccharomyces cerevisiae. PLOS Genetics, 1, e5. https://doi.org/10.1371/journal.pgen.0010005 [5] Ropars J, Rodríguez de la Vega RC, López-Villavicencio M, Gouzy J, Sallet E, Dumas É, Lacoste S, Debuchy R, Dupont J, Branca A, Giraud T (2015) Adaptive Horizontal Gene Transfers between Multiple Cheese-Associated Fungi. Current Biology, 25, 2562–2569. https://doi.org/10.1016/j.cub.2015.08.025 [6] Dumas E, Feurtey A, Rodríguez de la Vega RC, Le Prieur S, Snirc A, Coton M, Thierry A, Coton E, Le Piver M, Roueyre D, Ropars J, Branca A, Giraud T (2020) Independent domestication events in the blue-cheese fungus Penicillium roqueforti. Molecular Ecology, 29, 2639–2660. https://doi.org/10.1111/mec.15359 [7] Ropars J, Didiot E, Rodríguez de la Vega RC, Bennetot B, Coton M, Poirier E, Coton E, Snirc A, Le Prieur S, Giraud T (2020) Domestication of the Emblematic White Cheese-Making Fungus Penicillium camemberti and Its Diversification into Two Varieties. Current Biology, 30, 4441-4453.e4. https://doi.org/10.1016/j.cub.2020.08.082 [8] Caron T, Piver ML, Péron A-C, Lieben P, Lavigne R, Brunel S, Roueyre D, Place M, Bonnarme P, Giraud T, Branca A, Landaud S, Chassard C (2021) Strong effect of Penicillium roqueforti populations on volatile and metabolic compounds responsible for aromas, flavor and texture in blue cheeses. International Journal of Food Microbiology, 354, 109174. https://doi.org/10.1016/j.ijfoodmicro.2021.109174 [9] Ropars J, Lo Y-C, Dumas E, Snirc A, Begerow D, Rollnik T, Lacoste S, Dupont J, Giraud T, López-Villavicencio M (2016) Fertility depression among cheese-making Penicillium roqueforti strains suggests degeneration during domestication. Evolution, 70, 2099–2109. https://doi.org/10.1111/evo.13015 [10] Michel E, Masson E, Bubbendorf S, Lapicque L, Nidelet T, Segond D, Guézenec S, Marlin T, Devillers H, Rué O, Onno B, Legrand J, Sicard D, Bakers TP (2022) Artisanal and farmer bread making practices differently shape fungal species community composition in French sourdoughs. bioRxiv, 679472, ver. 6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/679472 [11] Vavilov NI, Vavylov MI, Dorofeev VF (1992) Origin and Geography of Cultivated Plants. Cambridge University Press. [12] Saslis-Lagoudakis CH, Clarke AC (2013) Ethnobiology: the missing link in ecology and evolution. Trends in Ecology & Evolution, 28, 67–68. https://doi.org/10.1016/j.tree.2012.10.017 [13] Thomas M, Demeulenaere E, Dawson JC, Khan AR, Galic N, Jouanne-Pin S, Remoue C, Bonneuil C, Goldringer I (2012) On-farm dynamic management of genetic diversity: the impact of seed diffusions and seed saving practices on a population-variety of bread wheat. Evolutionary Applications, 5, 779–795. https://doi.org/10.1111/j.1752-4571.2012.00257.x [14] Demeulenaere É, Lagrola M (2021) Des indicateurs pour accompagner “ les éleveurs de microbes” : Une communauté épistémique face au problème des laits “ paucimicrobiens ” dans la production fromagère au lait cru (1995-2015). Revue d’anthropologie des connaissances, 15. http://journals.openedition.org/rac/24953 | Artisanal and farmers bread making practices differently shape fungal species community composition in French sourdoughs | Elisa Michel, Estelle Masson, Sandrine Bubbendorf, Leocadie Lapicque, Thibault Nidelet, Diego Segond, Stephane Guezenec, Therese Marlin, Hugo deVillers, Olivier Rue, Bernard Onno, Judith Legrand, Delphine Sicard | <p style="text-align: justify;">Preserving microbial diversity in food systems is one of the many challenges to be met to achieve food security and quality. Although industrialization led to the selection and spread of specific fermenting microbia... | | Adaptation, Evolutionary Applications, Evolutionary Ecology | Tatiana Giraud | 2022-01-27 14:53:08 | ||
24 Oct 2019
Testing host-plant driven speciation in phytophagous insects : a phylogenetic perspectivePhylogenetic approaches for reconstructing macroevolutionary scenarios of phytophagous insect diversificationRecommended by Hervé Sauquet based on reviews by Brian O'Meara and 1 anonymous reviewerPlant-animal interactions have long been identified as a major driving force in evolution. However, only in the last two decades have rigorous macroevolutionary studies of the topic been made possible, thanks to the increasing availability of densely sampled molecular phylogenies and the substantial development of comparative methods. In this extensive and thoughtful perspective [1], Jousselin and Elias thoroughly review current hypotheses, data, and available macroevolutionary methods to understand how plant-insect interactions may have shaped the diversification of phytophagous insects. First, the authors review three main hypotheses that have been proposed to lead to host-plant driven speciation in phytophagous insects: the ‘escape and radiate’, ‘oscillation’, and ‘musical chairs’ scenarios, each with their own set of predictions. Jousselin and Elias then synthesize a vast core of recent studies on different clades of insects, where explicit phylogenetic approaches have been used. In doing so, they highlight heterogeneity in both the methods being used and predictions being tested across these studies and warn against the risk of subjective interpretation of the results. Lastly, they advocate for standardization of phylogenetic approaches and propose a series of simple tests for the predictions of host-driven speciation scenarios, including the characterization of host-plant range history and host breadth history, and diversification rate analyses. This helpful review will likely become a new point of reference in the field and undoubtedly help many researchers formalize and frame questions of plant-insect diversification in future studies of phytophagous insects. References [1] Jousselin, E., Elias, M. (2019). Testing Host-Plant Driven Speciation in Phytophagous Insects: A Phylogenetic Perspective. arXiv, 1910.09510, ver. 1 peer-reviewed and recommended by PCI Evol Biol. https://arxiv.org/abs/1910.09510v1 | Testing host-plant driven speciation in phytophagous insects : a phylogenetic perspective | Emmanuelle Jousselin, Marianne Elias | During the last two decades, ecological speciation has been a major research theme in evolutionary biology. Ecological speciation occurs when reproductive isolation between populations evolves as a result of niche differentiation. Phytophagous ins... | | Macroevolution, Phylogenetics / Phylogenomics, Speciation, Species interactions | Hervé Sauquet | 2019-02-25 17:31:33 | ||
06 Apr 2021
How robust are cross-population signatures of polygenic adaptation in humans?Be careful when studying selection based on polygenic score overdispersionRecommended by Torsten Günther based on reviews by Lawrence Uricchio, Mashaal Sohail, Barbara Bitarello and 1 anonymous reviewer
The advent of genome-wide association studies (GWAS) has been a great promise for our understanding of the connection between genotype and phenotype. Today, the NHGRI-EBI GWAS catalog contains 251,401 associations from 4,961 studies (1). This wealth of studies has also generated interest to use the summary statistics beyond the few top hits in order to make predictions for individuals without known phenotype, e.g. to predict polygenic risk scores or to study polygenic selection by comparing different groups. For instance, polygenic selection acting on the most studied polygenic trait, height, has been subject to multiple studies during the past decade (e.g. 2–6). They detected north-south gradients in Europe which were consistent with expectations. However, their GWAS summary statistics were based on the GIANT consortium data set, a meta-analysis of GWAS conducted in different European cohorts (7,8). The availability of large data sets with less stratification such as the UK Biobank (9) has led to a re-evaluation of those results. The nature of the GIANT consortium data set was realized to represent a potential problem for studies of polygenic adaptation which led several of the authors of the original articles to caution against the interpretations of polygenic selection on height (10,11). This was a great example on how the scientific community assessed their own earlier results in a critical way as more data became available. At the same time it left the question whether there is detectable polygenic selection separating populations more open than ever. Generally, recent years have seen several articles critically assessing the portability of GWAS results and risk score predictions to other populations (12–14). Refoyo-Martínez et al. (15) are now presenting a systematic assessment on the robustness of cross-population signatures of polygenic adaptation in humans. They compiled GWAS results for complex traits which have been studied in more than one cohort and then use allele frequencies from the 1000 Genomes Project data (16) set to detect signals of polygenic score overdispersion. As the source for the allele frequencies is kept the same across all tests, differences between the signals must be caused by the underlying GWAS. The results are concerning as the level of overdispersion largely depends on the choice of GWAS cohort. Cohorts with homogenous ancestries show little to no overdispersion compared to cohorts of mixed ancestries such as meta-analyses. It appears that the meta-analyses fail to fully account for stratification in their data sets. The authors based most of their analyses on the heavily studied trait height. Additionally, they use educational attainment (measured as the number of school years of an individual) as an example. This choice was due to the potential over- or misinterpretation of results by the media, the general public and by far right hate groups. Such traits are potentially confounded by unaccounted cultural and socio-economic factors. Showing that previous results about polygenic selection on educational attainment are not robust is an important result that needs to be communicated well. This forms a great example for everyone working in human genomics. We need to be aware that our results can sometimes be misinterpreted. And we need to make an effort to write our papers and communicate our results in a way that is honest about the limitations of our research and that prevents the misuse of our results by hate groups. This article represents an important contribution to the field. It is cruicial to be aware of potential methodological biases and technical artifacts. Future studies of polygenic adaptation need to be cautious with their interpretations of polygenic score overdispersion. A recommendation would be to use GWAS results obtained in homogenous cohorts. But even if different biobank-scale cohorts of homogeneous ancestry are employed, there will always be some remaining risk of unaccounted stratification. These conclusions may seem sobering but they are part of the scientific process. We need additional controls and new, different methods than polygenic score overdispersion for assessing polygenic selection. Last year also saw the presentation of a novel approach using sequence data and GWAS summary statistics to detect directional selection on a polygenic trait (17). This new method appears to be robust to bias stemming from stratification in the GWAS cohort as well as other confounding factors. Such new developments show light at the end of the tunnel for the use of GWAS summary statistics in the study of polygenic adaptation. References 1. Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Research. 2019 Jan 8;47(D1):D1005–12. doi: https://doi.org/10.1093/nar/gky1120 2. Turchin MC, Chiang CW, Palmer CD, Sankararaman S, Reich D, Hirschhorn JN. Evidence of widespread selection on standing variation in Europe at height-associated SNPs. Nature Genetics. 2012 Sep;44(9):1015–9. doi: https://doi.org/10.1038/ng.2368 3. Berg JJ, Coop G. A Population Genetic Signal of Polygenic Adaptation. PLOS Genetics. 2014 Aug 7;10(8):e1004412. doi: https://doi.org/10.1371/journal.pgen.1004412 4. Robinson MR, Hemani G, Medina-Gomez C, Mezzavilla M, Esko T, Shakhbazov K, et al. Population genetic differentiation of height and body mass index across Europe. Nature Genetics. 2015 Nov;47(11):1357–62. doi: https://doi.org/10.1038/ng.3401 5. Mathieson I, Lazaridis I, Rohland N, Mallick S, Patterson N, Roodenberg SA, et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015 Dec;528(7583):499–503. doi: https://doi.org/10.1038/nature16152 6. Racimo F, Berg JJ, Pickrell JK. Detecting polygenic adaptation in admixture graphs. Genetics. 2018. Arp;208(4):1565–1584. doi: https://doi.org/10.1534/genetics.117.300489 7. Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010 Oct;467(7317):832–8. doi: https://doi.org/10.1038/nature09410 8. Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014 Nov;46(11):1173–86. doi: https://doi.org/10.1038/ng.3097 9. Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018 Oct;562(7726):203–9. doi: https://doi.org/10.1038/s41586-018-0579-z 10. Berg JJ, Harpak A, Sinnott-Armstrong N, Joergensen AM, Mostafavi H, Field Y, et al. Reduced signal for polygenic adaptation of height in UK Biobank. eLife. 2019 Mar 21;8:e39725. doi: https://doi.org/10.7554/eLife.39725 11. Sohail M, Maier RM, Ganna A, Bloemendal A, Martin AR, Turchin MC, et al. Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. eLife. 2019 Mar 21;8:e39702. doi: https://doi.org/10.7554/eLife.39702 12. Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nature Genetics. 2019 Apr;51(4):584–91. doi: https://doi.org/10.1038/s41588-019-0379-x 13. Bitarello BD, Mathieson I. Polygenic Scores for Height in Admixed Populations. G3: Genes, Genomes, Genetics. 2020 Nov 1;10(11):4027–36. doi: https://doi.org/10.1534/g3.120.401658 14. Uricchio LH, Kitano HC, Gusev A, Zaitlen NA. An evolutionary compass for detecting signals of polygenic selection and mutational bias. Evolution Letters. 2019;3(1):69–79. doi: https://doi.org/10.1002/evl3.97 15. Refoyo-Martínez A, Liu S, Jørgensen AM, Jin X, Albrechtsen A, Martin AR, Racimo F. How robust are cross-population signatures of polygenic adaptation in humans? bioRxiv, 2021, 2020.07.13.200030, version 5 peer-reviewed and recommended by Peer community in Evolutionary Biology. doi: https://doi.org/10.1101/2020.07.13.200030 16. Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, et al. A global reference for human genetic variation. Nature. 2015 Sep 30;526(7571):68–74. doi: https://doi.org/10.1038/nature15393 17. Stern AJ, Speidel L, Zaitlen NA, Nielsen R. Disentangling selection on genetically correlated polygenic traits using whole-genome genealogies. bioRxiv. 2020 May 8;2020.05.07.083402. doi: https://doi.org/10.1101/2020.05.07.083402 | How robust are cross-population signatures of polygenic adaptation in humans? | Alba Refoyo-Martínez, Siyang Liu, Anja Moltke Jørgensen, Xin Jin, Anders Albrechtsen, Alicia R. Martin, Fernando Racimo | <p>Over the past decade, summary statistics from genome-wide association studies (GWASs) have been used to detect and quantify polygenic adaptation in humans. Several studies have reported signatures of natural selection at sets of SNPs associated... | | Bioinformatics & Computational Biology, Genetic conflicts, Human Evolution, Population Genetics / Genomics | Torsten Günther | 2020-08-14 15:06:54 | ||
08 Aug 2018

Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutationsInbreeding compensates for reduced sexual selection in purging deleterious mutationsRecommended by Charles Baer based on reviews by 2 anonymous reviewersTwo evolutionary processes have been shown in theory to enhance the effects of natural selection in purging deleterious mutations from a population (here ""natural"" selection is defined as ""selection other than sexual selection""). First, inbreeding, especially self-fertilization, facilitates the removal of deleterious recessive alleles, the effects of which are largely hidden from selection in heterozygotes when mating is random. Second, sexual selection can facilitate the removal of deleterious alleles of arbitrary dominance, with little or no demographic cost, provided that deleterious effects are greater in males than in females (""genic capture""). Inbreeding (especially selfing) and sexual selection are often negatively correlated in nature. Empirical tests of the role of sexual selection in purging deleterious mutations have been inconsistent, potentially due to the positive relationship between sexual selection and intersexual genetic conflict. References [1] Noël, E., Fruitet, E., Lelaurin, D., Bonel, N., Segard, A., Sarda, V., Jarne, P., & David P. (2018). Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations. bioRxiv, 273367, ver. 3 recommended and peer-reviewed by PCI Evol Biol. doi: 10.1101/273367 | Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations | E. Noël, E. Fruitet, D. Lelaurin, N. Bonel, A. Ségard, V. Sarda, P. Jarne and P. David | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (https://dx.doi.org/10.24072/pci.evolbiol.100055). Theory and empirical data showed that two processes can boost selection against deleterious mutations, ... | | Adaptation, Experimental Evolution, Reproduction and Sex, Sexual Selection | Charles Baer | Anonymous | 2018-03-01 08:12:37 | |
20 May 2020
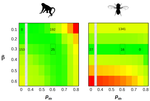
How much does Ne vary among species?Further questions on the meaning of effective population sizeRecommended by Martin Lascoux based on reviews by 3 anonymous reviewersIn spite of its name, the effective population size, Ne, has a complex and often distant relationship to census population size, as we usually understand it. In truth, it is primarily an abstract concept aimed at measuring the amount of genetic drift occurring in a population at any given time. The standard way to model random genetic drift in population genetics is the Wright-Fisher model and, with a few exceptions, definitions of the effective population size stems from it: “a certain model has effective population size, Ne, if some characteristic of the model has the same value as the corresponding characteristic for the simple Wright-Fisher model whose actual size is Ne” (Ewens 2004). Since Sewall Wright introduced the concept of effective population size in 1931 (Wright 1931), it has flourished and there are today numerous definitions of it depending on the process being examined (genetic diversity, loss of alleles, efficacy of selection) and the characteristic of the model that is considered. These different definitions of the effective population size were generally introduced to address specific aspects of the evolutionary process. One aspect that has been hotly debated since the first estimates of genetic diversity in natural populations were published is the so-called Lewontin’s paradox (1974). Lewontin noted that the observed variation in heterozygosity across species was much smaller than one would expect from the neutral expectations calculated with the actual size of the species. References Brandvain Y, Wright SI (2016) The Limits of Natural Selection in a Nonequilibrium World. Trends in Genetics, 32, 201–210. doi: 10.1016/j.tig.2016.01.004 | How much does Ne vary among species? | Nicolas Galtier, Marjolaine Rousselle | <p>Genetic drift is an important evolutionary force of strength inversely proportional to *Ne*, the effective population size. The impact of drift on genome diversity and evolution is known to vary among species, but quantifying this effect is a d... | | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Martin Lascoux | 2019-12-08 00:11:00 |
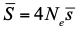 , where Ne is the effective population size and
, where Ne is the effective population size and  is the mean fitness effect of non-synonymous mutations. Assuming further that distinct species share a common DFE and therefore a common
is the mean fitness effect of non-synonymous mutations. Assuming further that distinct species share a common DFE and therefore a common  . Applying their newly developed approach to various datasets they conclude that the power of drift varies by a factor of at least 500 between large-Ne (Drosophila) and small-Ne species (H. sapiens). This is an order of magnitude larger than what would be obtained by comparing estimates of the variation in neutral diversity. Hence the proposed approach seems to have gone some way in making Lewontin’s paradox less paradoxical. But, perhaps more importantly, as the authors tersely point out at the end of the abstract their results further questions the meaning of Ne parameters in population genetics. And arguably this could well be the most important contribution of their study and something that is badly needed.
. Applying their newly developed approach to various datasets they conclude that the power of drift varies by a factor of at least 500 between large-Ne (Drosophila) and small-Ne species (H. sapiens). This is an order of magnitude larger than what would be obtained by comparing estimates of the variation in neutral diversity. Hence the proposed approach seems to have gone some way in making Lewontin’s paradox less paradoxical. But, perhaps more importantly, as the authors tersely point out at the end of the abstract their results further questions the meaning of Ne parameters in population genetics. And arguably this could well be the most important contribution of their study and something that is badly needed. 



