Y chromosome makes fruit flies die younger
Recommended by Gabriel Marais, Jean-François Lemaitre and Cristina Vieira
In most animal species, males and females display distinct survival prospect, a phenomenon known as sex gap in longevity (SGL, Marais et al. 2018). The study of SGLs is crucial not only for having a full picture of the causes underlying organisms’ health, aging and death but also to initiate the development of sex-specific anti-aging interventions in humans (Austad and Bartke 2015). Three non-mutually evolutionary causes have been proposed to underlie SGLs (Marais et al. 2018). First, SGLs could be the consequences of sex-differences in life history strategies. For example, evolving dimorphic traits (e.g. body size, ornaments or armaments) may imply unequal physiological costs (e.g. developmental, maintenance) between the sexes and this may result in differences in longevity and aging. Second, mitochondria are usually transmitted by the mother and thus selection is blind to mitochondrial deleterious mutations affecting only males. Such mutations can freely accumulate in the mitochondrial genome and may reduce male longevity, a phenomenon called the mother’s curse (Frank and Hurst 1996). Third, in species with sex chromosomes, all recessive deleterious mutations will be expressed on the single X chromosome in XY males and may reduce their longevity (the unguarded X effect). In addition, the numerous transposable elements (TEs) on the Y chromosome may affect aging. TE activity is normally repressed by epigenetic regulation (DNA methylation, histone modifications and small RNAs). However, it is known that this regulation is disrupted with increasing age. Because of the TE-rich Y chromosome, more TEs may become active in old males than in old females, generating more somatic mutations, accelerating aging and reducing longevity in males (the toxic Y effect, Marais et al. 2018).
The relative contributions of these different effects to SGLs remain unknown. Sex-differences in life history strategies have been considered as the most important cause of SGLs for long (Tidière et al. 2015) but this effect remain equivocal (Lemaître et al. 2020) and cannot explain alone the diversity of patterns observed across species (Marais et al. 2018). Similarly, while studies in Drosophila and humans have shown that the mother’s curse contributes to SGLs in those organisms (e.g. Milot et al. 2007), its contribution may not be strong. Recently, two large-scale comparative analyses have shown that in species with XY chromosomes males show a shorter lifespan compared to females, while in species with ZW chromosomes (a system in which the female are the heterogametic sex and are ZW, and the males ZZ) the opposite pattern is observed (Pipoly et al. 2015; Xirocostas et al. 2020). Apart from these correlational studies, very little experimental tests of the effect of sex chromosomes on longevity have been conducted. In Drosophila, the evidence suggests that the unguarded X effect does not contribute to SGLs (Brengdahl et al. 2018). Whether a toxic Y effect exists in this species was unknown.
In a very elegant study, Brown et al. (2020) provided strong evidence for such a toxic Y effect in Drosophila melanogaster. First, they checked that in the D. melanogaster strain that they were studying (Canton-S), males were indeed dying younger than females. They also confirmed that in this strain, as in others, the male genomes include more repeats and heterochromatin than the female ones using cytometry. A careful analysis of the heterochromatin (using H3K9me2, a repressive histone modification typical of heterochromatin, as a proxy) in old flies revealed that heterochromatin loss was much more important in males than in females, in particular on the Y chromosome (but also to a lesser extent at the pericentromric regions of the autosomes). This change in heterochromatin had two outputs, they found. First, the expression of the genes in those regions was affected. They highlighted that many of such genes are involved in immunity and regulation with a potential impact on longevity. Second, they found a striking TE reactivation. These two effects were stronger in males. While females showed clear reactivation of 6 TEs, with the total fraction of repeats in the transcriptome going from 2% (young females) to 4.6% (old females), males experienced the reactivation of 32 TEs, with the total fraction of repeats in the transcriptome going from 1.6% (young males) to 5.8% (old males). It appeared that most of these TEs are Y-linked. And when focusing on Y-linked repeats, they found that 32 Y-linked TEs became upregulated during male aging and the fraction of Y-linked TEs in the transcriptome increased ninefold.
All these observations clearly suggested that male longevity was decreased because of a toxic Y effect. To really uncover a causal relationship between having a Y chromosome and shorter longevity, Brown et al. (2020) artificially produced flies with atypical karyotypes: X0 males, XXY females and XYY males. This is very interesting as they could uncouple the effect of the phenotypical sex (being male or female) and having a Y chromosome or not, as in fruit flies sex is determined not by the Y chromosome but by the X/autosome ratio. Their results are striking. They found that longevity of the X0 males was the highest (higher than XX females in fact), and that of the XYY males the lowest. Females XXY had intermediate longevities. Importantly, this was found to be robust to genomic background as results were the same using crosses from different strains. When analysing TEs of these flies, they found a particularly strong expression of the Y-linked TEs in old XXY and XYY flies. Interestingly, in young XXY and XYY flies Y-linked TEs expression was also strong, suggesting the chromatin regulation of the Y chromosome is disrupted in these flies.
This work points to the idea that SGLs in D. melanogaster are mainly explained by the toxic Y effect. The molecular details however remain to be elucidated. The effect of the Y chromosome on aging might be more complex than envisioned in the toxic Y model presented above. Brown et al. (2020) indeed found that heterochromatin loss was globally faster in males, both at the Y chromosome and the autosomes. The organisation of the nucleus, in particular of the nucleolus, which is involved in heterochromatin maintenance, involves the sex chromosomes in D. melanogaster as discussed in the paper, and may explain this observation. The epigenetic status of the Y chromosome is known to affect that of all the autosomes in Drosophila (Lemos et al. 2008). Also, in Brown et al. (2020) most of the work (in particular the genomic part) has been done on Canton-S. Only D. melanogaster was studied but limited data suggest different Drosophila species may have different SGLs. The TE analysis is known to be tricky, different tools to analyse TE expression exist (e.g. Lerat et al. 2017; Lanciano and Cristofari 2020). Future work should focus on testing the toxic Y effect on other D. melanogaster strains and other Drosophila species, using different tools to study TE expression, and on dissecting the molecular details of the toxic Y effect.
References
Austad, S. N., and Bartke, A. (2015). Sex differences in longevity and in responses to anti-aging interventions: A Mini-Review. Gerontology, 62(1), 40–46. 10.1159/000381472
Brengdahl, M., Kimber, C. M., Maguire-Baxter, J., and Friberg, U. (2018). Sex differences in life span: Females homozygous for the X chromosome do not suffer the shorter life span predicted by the unguarded X hypothesis. Evolution; international journal of organic evolution, 72(3), 568–577. 10.1111/evo.13434
Brown, E. J., Nguyen, A. H., and Bachtrog, D. (2020). The Y chromosome may contribute to sex-specific ageing in Drosophila. Nature ecology and evolution, 4(6), 853–862. 10.1038/s41559-020-1179-5 or preprint link on bioRxiv
Frank, S. A., and Hurst, L. D. (1996). Mitochondria and male disease. Nature, 383(6597), 224. 10.1038/383224a0
Lanciano, S., and Cristofari, G. (2020). Measuring and interpreting transposable element expression. Nature reviews. Genetics, 10.1038/s41576-020-0251-y. Advance online publication. 10.1038/s41576-020-0251-y
Lemaître, J. F., Ronget, V., Tidière, M., Allainé, D., Berger, V., Cohas, A., Colchero, F., Conde, D. A., Garratt, M., Liker, A., Marais, G., Scheuerlein, A., Székely, T., and Gaillard, J. M. (2020). Sex differences in adult lifespan and aging rates of mortality across wild mammals. Proceedings of the National Academy of Sciences of the United States of America, 117(15), 8546–8553. 10.1073/pnas.1911999117
Lemos, B., Araripe, L. O., and Hartl, D. L. (2008). Polymorphic Y chromosomes harbor cryptic variation with manifold functional consequences. Science (New York, N.Y.), 319(5859), 91–93. 10.1126/science.1148861
Lerat, E., Fablet, M., Modolo, L., Lopez-Maestre, H., and Vieira, C. (2017). TEtools facilitates big data expression analysis of transposable elements and reveals an antagonism between their activity and that of piRNA genes. Nucleic acids research, 45(4), e17. 10.1093/nar/gkw953
Marais, G., Gaillard, J. M., Vieira, C., Plotton, I., Sanlaville, D., Gueyffier, F., and Lemaitre, J. F. (2018). Sex gap in aging and longevity: can sex chromosomes play a role?. Biology of sex differences, 9(1), 33. 10.1186/s13293-018-0181-y
Milot, E., Moreau, C., Gagnon, A., Cohen, A. A., Brais, B., and Labuda, D. (2017). Mother's curse neutralizes natural selection against a human genetic disease over three centuries. Nature ecology and evolution, 1(9), 1400–1406. 10.1038/s41559-017-0276-6
Pipoly, I., Bókony, V., Kirkpatrick, M., Donald, P. F., Székely, T., and Liker, A. (2015). The genetic sex-determination system predicts adult sex ratios in tetrapods. Nature, 527(7576), 91–94. 10.1038/nature15380
Tidière, M., Gaillard, J. M., Müller, D. W., Lackey, L. B., Gimenez, O., Clauss, M., and Lemaître, J. F. (2015). Does sexual selection shape sex differences in longevity and senescence patterns across vertebrates? A review and new insights from captive ruminants. Evolution; international journal of organic evolution, 69(12), 3123–3140. 10.1111/evo.12801
Xirocostas, Z. A., Everingham, S. E., and Moles, A. T. (2020). The sex with the reduced sex chromosome dies earlier: a comparison across the tree of life. Biology letters, 16(3), 20190867. 10.1098/rsbl.2019.0867
| The Y chromosome may contribute to sex-specific ageing in Drosophila | Emily J Brown, Alison H Nguyen, Doris Bachtrog | <p>Heterochromatin suppresses repetitive DNA, and a loss of heterochromatin has been observed in aged cells of several species, including humans and *Drosophila*. Males often contain substantially more heterochromatic DNA than females, due to the ... |  | Bioinformatics & Computational Biology, Expression Studies, Genetic conflicts, Genome Evolution, Genotype-Phenotype, Molecular Evolution, Reproduction and Sex | Gabriel Marais | | 2020-07-28 15:06:18 | View |
The insertion of a mitochondrial selfish element into the nuclear genome and its consequences
Julien Y. Dutheil, Karin Münch, Klaas Schotanus, Eva H. Stukenbrock and Regine Kahmann
https://doi.org/10.1101/787044
Some evolutionary insights into an accidental homing endonuclease passage from mitochondria to the nucleus
Recommended by Sylvain Charlat based on reviews by Jan Engelstaedter and Yannick Wurm
Not all genetic elements composing genomes are there for the benefit of their carrier. Many have no consequences on fitness, or too mild ones to be eliminated by selection, and thus stem from neutral processes. Many others are indeed the product of selection, but one acting at a different level, increasing the fitness of some elements of the genome only, at the expense of the “organism” as a whole. These can be called selfish genetic elements, and come into a wide variety of flavours [1], illustrating many possible means to cheat with “fair” reproductive processes such as meiosis, and thus get overrepresented in the offspring of their hosts. Producing copies of itself through transposition is one such strategy; a very successful one indeed, explaining a large part of the genomic content of many organisms. Killing non carrier gametes following meiosis in heterozygous carriers is another one. Less know and less common is the ability of some elements to turn heterozygous carriers into homozygous ones, that will thus transmit the selfish elements to all offspring instead of half. This is achieved by nucleic sequences encoding so-called “Homing endonucleases” (HEs). These proteins tend to induce double strand breaks of DNA specifically in regions homologous to their own insertion sites. The recombination machinery is such that the intact homologous region, that is, the one carrying the HE sequence, is then used as a template for the reparation of the break, resulting in the effective conversion of a non-carrier allele into a carrier allele. Such elements can also occur in the mitochondrial genomes of organisms where mitochondria are not strictly transmitted by one parent only, offering mitochondrial HEs some opportunities for “homing” into new non carrier genomes. This is the case in yeasts, where HEs were first reported [2,3].
In this new study, based on genomic experimental data from the fungal maize pathogen Ustilago maydis, Julien Dutheil and colleagues [4] document one possible evolutionary pathway for which little evidence existed before: the passage of a mitochondrial HE into the nuclear genome. The GC content of this region leaves little doubt on its mitochondrial origin, and homologs can indeed be found in the mitochondrial genomes of close relatives. Strangely enough, U. maydis itself does not appear to carry this selfish element in its own mitochondria, suggesting it may have been acquired from a different species, or be subject to a sufficiently rapid turnover to have been recently lost.
Many elements of the story uncovered by this study remain mysterious. How, in the first place, was this HE gene inserted in a nuclear genomic region that shows no apparent homology with its original insertion site, making typical “homing” a not-so-likely explanation? This question may in fact be generalised to many HE systems: is the first insertion into a homing site always the product of a typical homing event, which implies the presence of an homologous template DNA fragment, or can HE genes insert through other means? But then, why specifically in regions that would be targeted by the nuclease they encode? What is the evolutionary fate of this newly inserted element? The new gene may well be on its way to pseudogenisation, as suggested by the truncation of its upper part, precluding its functioning as a HE, and the lack of evidence of selective constraints through dN/dS analysis; but the mutation generated by the insertion event may have phenotypic implications, possibly through the partial truncation of another gene, encoding a helicase. How old is this insertion? The fact that it has accumulated some mutations makes a very recent event rather unlikely, but this insertion has been detected in only one isolate of U. maydis, suggesting it is not so frequent in natural populations.
Whatever the answers to these open questions, that will hopefully be addressed by further work on this system, the present study has revealed that horizontal transmission enlarges the scope of possible evolutionary consequences of HE genes, that may move not only between mitochondrial genomes, but also occasionally into a nucleus.
References
[1] Burt, A., and Trivers, R. (2006). Genes in Conflict: The Biology of Selfish Genetic Elements. Belknap Press.
[2] Coen, D., Deutch, J., Netter, P., Petrochillo, E., and Slonimski, P. (1970). Mitochondrial genetics. I. Methodology and phenomenology. Symposia of the Society for Experimental Biology, 24, 449-496.
[3] Colleaux, L., D’Auriol, L., Betermier, M., Cottarel, G., Jacquier, A., Galibert, F., and Dujon, B. (1986). Universal code equivalent of a yeast mitochondrial intron reading frame is expressed into E. coli as a specific double strand endonuclease. Cell, 44, 521–533. doi: 10.1016/0092-8674(86)90262-X
[4] Dutheil, J. Y., Münch, K., Schotanus, K., Stukenbrock, E. H., and Kahmann, R. (2020). The insertion of a mitochondrial selfish element into the nuclear genome and its consequences. bioRxiv, 787044, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/787044
| The insertion of a mitochondrial selfish element into the nuclear genome and its consequences | Julien Y. Dutheil, Karin Münch, Klaas Schotanus, Eva H. Stukenbrock and Regine Kahmann | <p>Homing endonucleases (HE) are enzymes capable of cutting DNA at highly specific target sequences, the repair of the generated double-strand break resulting in the insertion of the HE-encoding gene ("homing" mechanism). HEs are present in all th... |  | Genome Evolution, Molecular Evolution | Sylvain Charlat | | 2019-09-30 20:34:23 | View |
Distribution of iridescent colours in hummingbird communities results from the interplay between selection for camouflage and communication
Hugo Gruson, Marianne Elias, Juan L. Parra, Christine Andraud, Serge Berthier, Claire Doutrelant, Doris Gomez
https://doi.org/10.1101/586362
Feathers iridescence sheds light on the assembly rules of humingbirds communities
Recommended by Sébastien Lavergne based on reviews by 2 anonymous reviewers
Ecology needs rules stipulating how species distributions and ecological communities should be assembled along environmental gradients, but few rules have yet emerged in the ecological literature. The search of ecogeographical rules governing the spatial variation of birds colours has recently known an upsurge of interest in the litterature [1]. Most studies have, however, looked at pigmentary colours and not structural colours (e.g. iridescence), although it is know that color perception by animals (both birds and their predators) can be strongly influenced by light diffraction causing iridescence patterns on feathers.
In the present study [2], the authors study ca. 190 ecological communities of hummingbirds as a function of their iridescent colors, in a large study zone spanning varied habitats across Ecuador. They show that colour composition of local hummingbirds communities are shaped by two main processes :
(i) phenotyping clustering of birds with similar dorsal colours, due to local selection of species with similar camouflages against predators (i.e. some sort of mimetic circles).
(ii) phenotypic overdispersion of birds with distinct facial and ventral colours, resulting from character displacement and limiting reproductive interference.
I found this second result particularly interesting because it adds to the mounting evidence that character displacement (also for songs or olfactory signaling) allow local coexistence between closely-related bird species once they have reached secondary sympatry. It is important to note that not all color patches though to be involved in sexual selection followed this overdispersion rule -- throat and crown color patches were not found overdispersed. This suggests that further investigation is needed to determine how color variation shape the structure of hummingbird communities, or bird communities in general.
Another notable quality of the present study is that it is making extensive use of museum specimens and thus shows that very innovative research can be performed with museum collections.
References
[1] Delhey, K. (2019). A review of Gloger’s rule, an ecogeographical rule of colour: definitions, interpretations and evidence. Biological Reviews, 94(4), 1294–1316. doi: 10.1111/brv.12503
[2] Gruson, H., Elias, M., Parra, J. L., Andraud, C., Berthier, S., Doutrelant, C., & Gomez, D. (2019). Distribution of iridescent colours in hummingbird communities results from the interplay between selection for camouflage and communication. BioRxiv, 586362, v5 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/586362
| Distribution of iridescent colours in hummingbird communities results from the interplay between selection for camouflage and communication | Hugo Gruson, Marianne Elias, Juan L. Parra, Christine Andraud, Serge Berthier, Claire Doutrelant, Doris Gomez | <p>Identification errors between closely related, co-occurring, species may lead to misdirected social interactions such as costly interbreeding or misdirected aggression. This selects for divergence in traits involved in species identification am... | 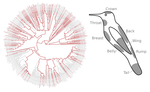 | Evolutionary Ecology, Macroevolution, Phylogeography & Biogeography, Sexual Selection, Species interactions | Sébastien Lavergne | | 2019-03-29 17:23:20 | View |
Evolution of selfing & lifespan 2.0
Recommended by Thomas Bataillon based on reviews by 2 anonymous reviewers
Flowering plants display a staggering diversity of both mating systems and life histories, ranging from almost exclusively selfers to obligate outcrossers, very short-lived annual herbs to super long lived trees. One pervasive pattern that has attracted considerable attention is the tight correlation that is found between mating systems and lifespan [1]. Until recently, theoretical explanations for these patterns have relied on static models exploring the consequences of several non-mutually exclusive important process: levels of inbreeding depression and ability to successfully were center stage. This make sense: successful colonization after long‐distance dispersal is far more likely to happen for self‐compatible than for self‐incompatible individuals in a sexually reproducing species. Furthermore, inbreeding depression (essentially a genetically driven phenomenon) and reproductive insurance are expected to shape the evolution of both mating system and lifespan.
But modelling jointly several processes and how their interplay to shape the evolution of a trait is challenging enough so models for the evolution of mating system tend invariably – for mathematical convenience and tractability – to fix lifespan [2].
However, comparative analysis of between species variations that map traits transitions among sister species in phylogenetic trees reveals a pervasive pattern: frequent transitions from a state outcrossing perennial to selfing annuals. This beg the question: is one transition triggering the other and if so, what comes first or are these transitions happening together?
In this work, Lesaffre and Billiard use a very sophisticated machinery developed by Kirkpatrick et al. [3] and consider a general class of so-called modifiers models [4]. They study jointly the evolution of life span and mating system. They do so by using models where different life stages are tracked with life stage having some (fixed for once) amount of inbreeding depression. Their paper is technically demanding, mixing analytics and computer simulations, and along the way generates several important findings that are expected to stimulate further empirical and theoretical studies: (1) pure selfing versus pure outcrossing is the expected stable evolutionary outcomes (despite observation that mixed mating systems can be regularly met in nature), (2) increasing life-span drastically reduces the scope for the evolution of selfing, conversely (3) transition to selfing will also select for shorter life span as a way to mitigate the cumulative effects of inbreeding depression on adult life stages.
As usual there is room for future work, in particular the authors’ model assumes fixed inbreeding depression in the different life stages and this highlights the need for models that explore how inbreeding depression, a pivotal quantity in these models, can itself be molded by both mating system and lifespan. A third-generation of models should be “soon” on the way!
References
[1] Grossenbacher D, Briscoe Runquist R, Goldberg EE, and Brandvain Y. (2015) Geographic range size is predicted by plant mating system. Ecology Letters 18, 706–713. doi: 10.1111/ele.12449
[2] Morgan MT, Schoen DJ, and Bataillon T. (1997) The evolution of self-fertilization in perennials. The American Naturalist 150, 618–638. doi: 10.1086/286085
[3] Kirkpatrick M, Johnson T, and Barton N. (2002) General models of multilocus evolution. Genetics 161, 1727–1750.
[4] Lesaffre, T, and Billiard S. (2019) The joint evolution of lifespan and self-fertilisation. bioRxiv, 420877, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/420877
| The joint evolution of lifespan and self-fertilisation | Thomas Lesaffre, Sylvain Billiard | <p>In Angiosperms, there exists a strong association between mating system and lifespan. Most self-fertilising species are short-lived and most predominant or obligate outcrossers are long-lived. This association is generally explained by the infl... |  | Evolutionary Theory, Life History, Reproduction and Sex | Thomas Bataillon | | 2018-09-19 10:03:51 | View |
Epistasis and the evolution of selfing
Recommended by Sylvain Gandon based on reviews by Nick Barton and 1 anonymous reviewer
The evolution of selfing results from a balance between multiple evolutionary forces. Selfing provides an "automatic advantage" due to the higher efficiency of selfers to transmit their genes via selfed and outcrossed offspring. Selfed offspring, however, may suffer from inbreeding depression. In principle the ultimate evolutionary outcome is easy to predict from the relative magnitude of these two evolutionary forces [1,2]. Yet, several studies explicitly taking into account the genetic architecture of inbreeding depression noted that these predictions are often too restrictive because selfing can evolve in a broader range of conditions [3,4].
The present work by Abu Awad and Roze [5] provides an analytic understanding of these results. Abu Awad and Roze analyse the evolution of selfing in a multilocus model where some loci are coding for selfing while others are under direct selection. The evolution of selfing depends on (i) the classical benefit of selfing (automatic advantage), (ii) the cost of selfing due to inbreeding depression, (iii) the association between the loci coding for selfing and the loci under direct selection (likely to be positive because selfing is expected to be found in better purged genetic backgrounds) and (iv) the association between the loci coding for selfing and the linkage between loci under selection (this final term depends on the magnitude and the type of epistasis). Because these last two terms depend on genetic associations they are expected to play in when selection is strong and recombination is small. These last two terms explain why selfing is evolving under a range of conditions which is broader than predicted by earlier theoretical models. The match between the approximations for the different terms acting on the evolution of selfing and individual based simulations (for different fitness landscapes) is very convincing. In particular, this analysis also yields new results on the effect of different types of epistasis on inbreeding depression.
Another remarkable and important feature of this work is its readability. The analysis of multilocus models rely on several steps and approximations that often result in overwhelmingly complex papers. Abu Awad and Roze’s paper [5] is dense but it provides a very clear and comprehensive presentation of the interplay between multiple evolutionary forces acting on the evolution of selfing.
References
[1] Holsinger, K. E., Feldman, M. W., and Christiansen, F. B. (1984). The evolution of self-fertilization in plants: a population genetic model. The American Naturalist, 124(3), 446-453. doi: 10.1086/284287
[2] Lande, R., and Schemske, D. W. (1985). The evolution of self‐fertilization and inbreeding depression in plants. I. Genetic models. Evolution, 39(1), 24-40. doi: 10.1111/j.1558-5646.1985.tb04077.x
[3] Charlesworth, D., Morgan, M. T., and Charlesworth, B. (1990). Inbreeding depression, genetic load, and the evolution of outcrossing rates in a multilocus system with no linkage. Evolution, 44(6), 1469-1489. doi: 10.1111/j.1558-5646.1990.tb03839.x
[4] Uyenoyama, M. K., and Waller, D. M. (1991). Coevolution of self-fertilization and inbreeding depression I. Mutation-selection balance at one and two loci. Theoretical population biology, 40(1), 14-46. doi: 10.1016/0040-5809(91)90045-H
[5] Abu Awad, D. and Roze, D. (2020). Epistasis, inbreeding depression and the evolution of self-fertilization. bioRxiv, 809814, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/809814
| Epistasis, inbreeding depression and the evolution of self-fertilization | Diala Abu Awad and Denis Roze | <p>Inbreeding depression resulting from partially recessive deleterious alleles is thought to be the main genetic factor preventing self-fertilizing mutants from spreading in outcrossing hermaphroditic populations. However, deleterious alleles may... |  | Evolutionary Theory, Quantitative Genetics, Reproduction and Sex | Sylvain Gandon | | 2019-10-18 09:29:41 | View |
Geographic variation in adult and embryonic desiccation tolerance in a terrestrial-breeding frog
Rudin-Bitterli, T, Evans, J. P. and Mitchell, N. J.
https://doi.org/10.1101/314351
Tough as old boots: amphibians from drier habitats are more resistant to desiccation, but less flexible at exploiting wet conditions
Recommended by Ben Phillips based on reviews by Juan Diego Gaitan-Espitia, Jennifer Nicole Lohr and 1 anonymous reviewer
Species everywhere are facing rapid climatic change, and we are increasingly asking whether populations will adapt, shift, or perish [1]. There is a growing realisation that, despite limited within-population genetic variation, many species exhibit substantial geographic variation in climate-relevant traits. This geographic variation might play an important role in facilitating adaptation to climate change [2,3].
Much of our understanding of geographic variation in climate-relevant traits comes from model organisms [e.g. 4]. But as our concern grows, we make larger efforts to understand geographic variation in non-model organisms also. If we understand what adaptive geographic variation exists within a species, we can make management decisions around targeted gene flow [5]. And as empirical examples accumulate, we can look for generalities that can inform management of unstudied species [e.g. 6,7]. Rudin-Bitterli’s paper [8] is an excellent contribution in this direction.
Rudin-Bitterli and her co-authors [8] sampled six frog populations distributed across a strong rainfall gradient. They then assayed these frogs and their offspring for a battery of fitness-relevant traits. The results clearly show patterns consistent with local adaptation to water availability, but they also reveal trade-offs. In their study, frogs from the driest source populations were resilient to the hydric environment: it didn’t really affect them very much whether they were raised in wet or dry environments. By contrast, frogs from wet source areas did better in wet environments, and they tended to do better in these wet environments than did animals from the dry-adapted populations. Thus, it appears that the resilience of the dry-adapted populations comes at a cost: frogs from these populations cannot ramp up performance in response to ideal (wet) conditions.
These data have been carefully and painstakingly collected, and they are important. They reveal not only important geographic variation in response to hydric stress (in a vertebrate), but they also adumbrate a more general trade-off: that the jack of all trades might be master of none. Specialist-generalist trade-offs are often argued (and regularly observed) to exist [e.g. 9,10], and here we see them arise in climate-relevant traits also. Thus, Rudin-Bitterli’s paper is an important piece of the empirical puzzle, and one that points to generalities important for both theory and management.
References
[1] Hoffmann, A. A., and Sgrò, C. M. (2011). Climate change and evolutionary adaptation. Nature, 470(7335), 479–485. doi: 10.1038/nature09670
[2] Aitken, S. N., and Whitlock, M. C. (2013). Assisted Gene Flow to Facilitate Local Adaptation to Climate Change. Annual Review of Ecology, Evolution, and Systematics, 44(1), 367–388. doi: 10.1146/annurev-ecolsys-110512-135747
[3] Kelly, E., and Phillips, B. L. (2016). Targeted gene flow for conservation. Conservation Biology, 30(2), 259–267. doi: 10.1111/cobi.12623
[4] Sgrò, C. M., Overgaard, J., Kristensen, T. N., Mitchell, K. A., Cockerell, F. E., and Hoffmann, A. A. (2010). A comprehensive assessment of geographic variation in heat tolerance and hardening capacity in populations of Drosophila melanogaster from eastern Australia. Journal of Evolutionary Biology, 23(11), 2484–2493. doi: 10.1111/j.1420-9101.2010.02110.x
[5] Macdonald, S. L., Llewelyn, J., and Phillips, B. L. (2018). Using connectivity to identify climatic drivers of local adaptation. Ecology Letters, 21(2), 207–216. doi: 10.1111/ele.12883
[6] Hoffmann, A. A., Chown, S. L., and Clusella‐Trullas, S. (2012). Upper thermal limits in terrestrial ectotherms: how constrained are they? Functional Ecology, 27(4), 934–949. doi: 10.1111/j.1365-2435.2012.02036.x
[7] Araújo, M. B., Ferri‐Yáñez, F., Bozinovic, F., Marquet, P. A., Valladares, F., and Chown, S. L. (2013). Heat freezes niche evolution. Ecology Letters, 16(9), 1206–1219. doi: 10.1111/ele.12155
[8] Rudin-Bitterli, T. S., Evans, J. P., and Mitchell, N. J. (2019). Geographic variation in adult and embryonic desiccation tolerance in a terrestrial-breeding frog. BioRxiv, 314351, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. doi: 10.1101/314351
[9] Kassen, R. (2002). The experimental evolution of specialists, generalists, and the maintenance of diversity. Journal of Evolutionary Biology, 15(2), 173–190. doi: 10.1046/j.1420-9101.2002.00377.x
[10] Angilletta, M. J. J. (2009). Thermal Adaptation: A theoretical and empirical synthesis. Oxford University Press, Oxford.
| Geographic variation in adult and embryonic desiccation tolerance in a terrestrial-breeding frog | Rudin-Bitterli, T, Evans, J. P. and Mitchell, N. J. | <p>Intra-specific variation in the ability of individuals to tolerate environmental perturbations is often neglected when considering the impacts of climate change. Yet this information is potentially crucial for mitigating any deleterious effects... |  | Adaptation, Evolutionary Applications, Evolutionary Ecology | Ben Phillips | | 2018-05-07 03:35:08 | View |
Does the seed fall far from the tree? Weak fine scale genetic structure in a continuous Scots pine population
Alina K. Niskanen, Sonja T. Kujala, Katri Kärkkäinen, Outi Savolainen, Tanja Pyhäjärvi
https://doi.org/10.1101/2023.06.16.545344
Weak spatial genetic structure in a large continuous Scots pine population – implications for conservation and breeding
Recommended by Myriam Heuertz based on reviews by Joachim Mergeay, Jean-Baptiste Ledoux and Roberta Loh based on reviews by Joachim Mergeay, Jean-Baptiste Ledoux and Roberta Loh
Spatial genetic structure, i.e. the non-random spatial distribution of genotypes, arises in populations because of different processes including spatially limited dispersal and selection. Knowledge on the spatial genetic structure of plant populations is important to assess biological parameters such as gene dispersal distances and the potential for local adaptations, as well as for applications in conservation management and breeding. In their work, Niskanen and colleagues demonstrate a multifaceted approach to characterise the spatial genetic structure in two replicate sites of a continuously distributed Scots pine population in South-Eastern Finland. They mapped and assessed the ages of 469 naturally regenerated adults and genotyped them using a SNP array which resulted in 157 325 filtered polymorphic SNPs. Their dataset is remarkably powerful because of the large numbers of both individuals and SNPs genotyped. This made it possible to characterise precisely the decay of genetic relatedness between individuals with spatial distance despite the extensive dispersal capacity of Scots pine through pollen, and ensuing expectations of an almost panmictic population.
The authors’ data analysis was particularly thorough. They demonstrated that two metrics of pairwise relatedness, the genomic relationship matrix (GRM, Yang et al. 2011) and the kinship coefficient (Loiselle et al. 1995) were strongly correlated and produced very similar inference of family relationships: >99% of pairs of individuals were unrelated, and the remainder exhibited 2nd (e.g., half-siblings) to 4th degree relatedness. Pairwise relatedness decayed with spatial distance which resulted in extremely weak but statistically significant spatial genetic structure in both sites, quantified as Sp=0.0005 and Sp=0.0008. These estimates are at least an order of magnitude lower than estimates in the literature obtained in more fragmented populations of the same species or in other conifers. Estimates of the neighbourhood size, the effective number of potentially mating individuals belonging to a within-population neighbourhood (Wright 1946), were relatively large with Nb=1680-3210 despite relatively short gene dispersal distances, σg = 36.5–71.3m, which illustrates the high effective density of the population.
The authors showed the implications of their findings for selection. The capacity for local adaptation depends on dispersal distances and the strength of the selection coefficient. In the study population, the authors inferred that local adaptation can only occur if environmental heterogeneity occurs over a distance larger than approximately one kilometre (or larger, if considering long-distance dispersal). Interestingly, in Scots pine, no local adaptation has been described on similar geographic scales, in contrast to some other European or Mediterranean conifers (Scotti et al. 2023).
The authors’ results are relevant for the management of conservation and breeding. They showed that related individuals occurred within sites only and that they shared a higher number of rare alleles than unrelated ones. Since rare alleles are enriched in new and recessive deleterious variants, selecting related individuals could have negative consequences in breeding programmes. The authors also showed, in their response to reviewers, that their powerful dataset was not suitable to obtain a robust estimate of effective population size, Ne, based on the linkage disequilibrium method (Do et al. 2014). This illustrated that the estimation of Ne used for genetic indicators supported in international conservation policy (Hoban et al. 2020, CBD 2022) remains challenging in large and continuous populations (see also Santo-del-Blanco et al. 2023, Gargiulo et al. 2024).
References
CBD (2022) Kunming-Montreal Global Biodiversity Framework. https://www.cbd.int/doc/decisions/cop-15/cop-15-dec-04-en.pdf
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014). NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne ) from genetic data. Molecular Ecology Resources 14: 209–214. https://doi.org/10.1111/1755-0998.12157
Gargiulo R, Decroocq V, González-Martínez SC, Paz-Vinas I, Aury JM, Kupin IL, Plomion C, Schmitt S, Scotti I, Heuertz M (2024) Estimation of contemporary effective population size in plant populations: limitations of genomic datasets. Evolutionary Applications, in press, https://doi.org/10.1101/2023.07.18.549323
Hoban S, Bruford M, D’Urban Jackson J, Lopes-Fernandes M, Heuertz M, Hohenlohe PA, Paz-Vinas I, et al. (2020) Genetic diversity targets and indicators in the CBD post-2020 Global Biodiversity Framework must be improved. Biological Conservation 248: 108654. https://doi.org/10.1016/j.biocon.2020.108654
Loiselle BA, Sork VL, Nason J & Graham C (1995) Spatial genetic structure of a tropical understorey shrub, Psychotria officinalis (Rubiaceae). American Journal of Botany 82: 1420–1425. https://doi.org/10.1002/j.1537-2197.1995.tb12679.x
Santos-del-Blanco L, Olsson S, Budde KB, Grivet D, González-Martínez SC, Alía R, Robledo-Arnuncio JJ (2022). On the feasibility of estimating contemporary effective population size (Ne) for genetic conservation and monitoring of forest trees. Biological Conservation 273: 109704. https://doi.org/10.1016/j.biocon.2022.109704
Scotti I, Lalagüe H, Oddou-Muratorio S, Scotti-Saintagne C, Ruiz Daniels R, Grivet D, et al. (2023) Common microgeographical selection patterns revealed in four European conifers. Molecular Ecology 32: 393-411. https://doi.org/10.1111/mec.16750
Wright S (1946) Isolation by distance under diverse systems of mating. Genetics 31: 39–59. https://doi.org/10.1093/genetics/31.1.39
Yang J, Lee SH, Goddard ME & Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. The American Journal of Human Genetics 88: 76–82. https://www.cell.com/ajhg/pdf/S0002-9297(10)00598-7.pdf
| Does the seed fall far from the tree? Weak fine scale genetic structure in a continuous Scots pine population | Alina K. Niskanen, Sonja T. Kujala, Katri Kärkkäinen, Outi Savolainen, Tanja Pyhäjärvi | <p>Knowledge of fine-scale spatial genetic structure, i.e., the distribution of genetic diversity at short distances, is important in evolutionary research and in practical applications such as conservation and breeding programs. In trees, related... | 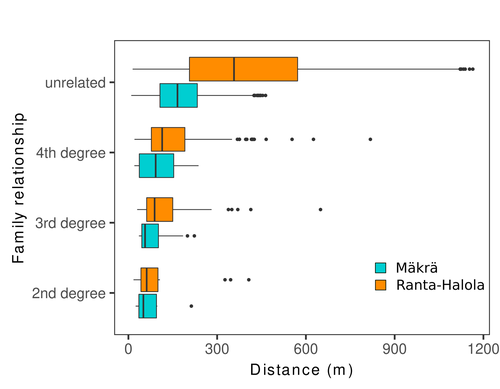 | Adaptation, Evolutionary Applications, Population Genetics / Genomics | Myriam Heuertz | Joachim Mergeay | 2023-06-27 21:57:28 | View |
Strong habitat and weak genetic effects shape the lifetime reproductive success in a wild clownfish population
Océane C. Salles, Glenn R. Almany, Michael L. Berumen, Geoffrey P. Jones, Pablo Saenz-Agudelo, Maya Srinivasan, Simon Thorrold, Benoit Pujol, Serge Planes
https://doi.org/10.5281/zenodo.3476529
Habitat variation of wild clownfish population shapes selfrecruitment more than genetic effects
Recommended by Philip Munday based on reviews by Juan Diego Gaitan-Espitia and Loeske Kruuk
Estimating the genetic and environmental components of variation in reproductive success is crucial to understanding the adaptive potential of populations to environmental change. To date, the heritability of lifetime reproductive success (fitness) has been estimated in a handful of wild animal population, mostly in mammals and birds, but has never been estimated for a marine species. The primary reason that such estimates are lacking in marine species is that most marine organisms have a dispersive larval phase, making it extraordinarily difficult to track the fate of offspring from one generation to the next.
In this study, Salles et al. [1] use an unprecedented 10 year data set for a wild population of orange clownfish (Amphiprion percula) to estimate the environmental, maternal and additive genetic components of life time reproductive success for the self-recruiting portion of the local population. Previous studies show that over 50% of juvenile clownfish recruiting to the population of clownfish at Kimbe Island (Kimbe Bay, PNG) are natal to the population. In other words, >50% of the juveniles recruiting to the population at Kimbe Island are offspring of parents from Kimbe Island. The identity and location of every adult clownfish in the Kimbe Island population was tracked over 10 years. At the same time newly recruiting juveniles were collected at regular intervals (biennially) and their parentage assigned with high confidence by 22 polymorphic microsatellite loci. Salles et al. then used a pedigree comprising 1735 individuals from up to 5 generations of clownfish at Kimbe Island to assess the contribution of every breeding pair of clownfish to self-recruitment within the local population. Because clownfish are site attached and live in close association with a host sea anemone, it was also possible to examine the contribution of reef location and host anemones species (either Heteractis magnifica or Stichodactyla gigantea) to reproductive success within the local population.
The study found that breeders from the eastern side of Kimbe Island, and mostly inhabiting S. gigantea sea anemones, produced more juveniles that recruited to the local population than breeders from other location around the island, or inhabiting H. magnifica. In fact, host anemone species and geographic location explained about 97% of the variance in reproductive success within the local population (i.e. excluding successful recruitment to other populations). By contrast, maternal and additive genetic effects explained only 1.9% and 1.3% of the variance, respectively. In other words, reef location and the species of host anemone inhabited had an overwhelming influence on the long-term contribution of breeding pairs of clownfish to replenishment of the local population. This overwhelming effect of the local habitat on reproductive success means that the population is potentially susceptible to rapid environmental changes - for example if S. giganta sea anemones are disproportionately susceptible to global warming, or reef habitats on the eastern side of the island are more susceptible to disturbance. By contrast, the small component of additive genetic variance in local reproductive success translated into low heritability and evolvability of lifetime reproductive success within the local population, as predicted by theory [2] and observed in some terrestrial species. Consequently, fitness would evolve slowly to environmental change.
Establishing the components of variation in fitness in a wild population of marine fishes is an astonishing achievement, made possible by the unprecedented long-term individual-level monitoring of the entire population of clownfish at Kimbe Island. A next step in this research would be to include other clownfish populations that are demographically and genetically connected to the Kimbe Island population through larval dispersal. It would be intriguing to establish the environmental, maternal and additive genetic components of reproductive success in the dispersing part of the Kimbe Island population, to see if this potentially differs among breeders who contribute more or less to replenishment within the local population.
References
[1] Salles, O. C., Almany, G. R., Berumen, M.L., Jones, G. P., Saenz-Agudelo, P., Srinivasan, M., Thorrold, S. R., Pujol, B., Planes, S. (2019). Strong habitat and weak genetic effects shape the lifetime reproductive success in a wild clownfish population. Zenodo, 3476529, ver. 3 peer-reviewed and recommended by Peer Community In Evolutionary Biology. doi: 10.5281/zenodo.3476529
[2] Fisher, R.A. (1930). The genetical theory of natural selection. Clarendon Press, Oxford, U.K.
| Strong habitat and weak genetic effects shape the lifetime reproductive success in a wild clownfish population | Océane C. Salles, Glenn R. Almany, Michael L. Berumen, Geoffrey P. Jones, Pablo Saenz-Agudelo, Maya Srinivasan, Simon Thorrold, Benoit Pujol, Serge Planes | <p>Lifetime reproductive success (LRS), the number of offspring an individual contributes to the next generation, is of fundamental importance in ecology and evolutionary biology. LRS may be influenced by environmental, maternal and additive genet... |  | Adaptation, Evolutionary Ecology, Life History, Quantitative Genetics | Philip Munday | | 2018-10-01 09:00:53 | View |
Environmental and fitness landscapes matter for the genetic basis of local adaptation
Recommended by Charles Mullon based on reviews by 2 anonymous reviewers
Natural landscapes are often composite, with spatial variation in environmental factors being the norm rather than exception. Adaptation to such variation is a major driver of diversity at all levels of biological organization, from genes to phenotypes, species and ultimately ecosystems. While natural selection favours traits that show a better fit to local conditions, the genomic response to such selection is not necessarily straightforward. This is because many quantitative traits are complex and the product of many loci, each with a small to moderate phenotypic contribution. Adapting to environmental challenges that occur in narrow ranges may thus prove difficult as each individual locus is easily swamped by alleles favoured across the rest of the population range.
To better understand whether and how evolution overcomes such a hurdle, Laroche and Lenormand [1] combine quantitative genetics and population genetic modelling to track genomic changes that underpin a trait whose fitness optimum differs between a certain spatial range, referred to as a “pocket”, and the rest of the habitat. As it turns out from their analysis, one critical and probably underappreciated factor in determining the type of genetic architecture that evolves is how fitness declines away from phenotypic optima. One classical and popular model of fitness landscape that relates trait value to reproductive success is Gaussian, whereby small trait variations away from the optimum result in even smaller variations in fitness. This facilitates local adaptation via the invasion of alleles of small effects as carriers inside the pocket show a better fit while those outside the pocket only suffer a weak fitness cost. By contrast, when the fitness landscape is more peaked around the optimum, for instance where the decline is linear, adaptation through weak effect alleles is less likely, requiring larger pockets that are less easily swamped by alleles selected in the rest of the range.
In addition to mathematically investigating the initial emergence of local adaptation, Laroche and Lenormand use computer simulations to look at its long-term maintenance. In principle, selection should favour a genetic architecture that consolidates the phenotype and increases its heritability, for instance by grouping several alleles of large effects close to one another on a chromosome to avoid being broken down by meiotic recombination. Whether or not this occurs also depends on the fitness landscape. When the landscape is Gaussian, the genetic architecture of the trait eventually consists of tightly linked alleles of large effects. The replacement of small effects by large effects loci is here again promoted by the slow fitness decline around the optimum. This is because any shift in architecture in an adapted population requires initially crossing a fitness valley. With a Gaussian landscape, this valley is shallow enough to be crossed, facilitated by a bit of genetic drift. By contrast, when fitness declines linearly around the optimum, genetic architecture is much less evolutionarily labile as any architecture change initially entails a fitness cost that is too high to bear.
Overall, Laroche and Lenormand provide a careful and thought-provoking analysis of a classical problem in population genetics. In addition to questioning some longstanding modelling assumptions, their results may help understand why differentiated populations are sometimes characterized by “genomic islands” of divergence, and sometimes not.
References
[1] Laroche F, Lenormand T (2022) The genetic architecture of local adaptation in a cline. bioRxiv, 2022.06.30.498280, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.06.30.498280
| The genetic architecture of local adaptation in a cline | Fabien Laroche, Thomas Lenormand | <p>Local adaptation is pervasive. It occurs whenever selection favors different phenotypes in different environments, provided that there is genetic variation for the corresponding traits and that the effect of selection is greater than the effect... | 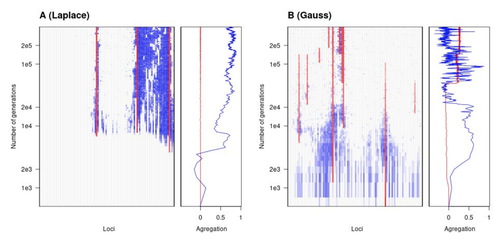 | Adaptation, Evolutionary Theory, Genome Evolution, Molecular Evolution, Population Genetics / Genomics, Quantitative Genetics | Charles Mullon | | 2022-07-07 08:46:47 | View |
Architectural traits constrain the evolution of unisexual flowers and sexual segregation within inflorescences: an interspecific approach
Rubén Torices, Ana Afonso, Arne A. Anderberg, José M. Gómez and Marcos Méndez
https://doi.org/10.1101/356147
Sometimes, sex is in the head
Recommended by Juan Arroyo based on reviews by 3 anonymous reviewers
Plants display an amazing diversity of reproductive strategies with and without sex. This diversity is particularly remarkable in flowering plants, as highlighted by Charles Darwin, who wrote several botanical books scrutinizing plant reproduction. One particularly influential work concerned floral variation [1]. Darwin recognized that flowers may present different forms within a single population, with or without sex specialization. The number of species concerned is small, but they display recurrent patterns, which made it possible for Darwin to invoke natural and sexual selection to explain them. Most of early evolutionary theory on the evolution of reproductive strategies was developed in the first half of the 20th century and was based on animals. However, the pioneering work by David Lloyd from the 1970s onwards excited interest in the diversity of plant sexual strategies as models for testing adaptive hypotheses and predicting reproductive outcomes [2]. The sex specialization of individual flowers and plants has since become one of the favorite topics of evolutionary biologists. However, attention has focused mostly on cases related to sex differentiation (dioecy and associated conditions [3]). Separate unisexual flower types on the same plant (monoecy and related cases, rendering the plant functionally hermaphroditic) have been much less studied, apart from their possible role in the evolution of dioecy [4] or their association with particular modes of pollination [5].
Two specific non-mutually exclusive hypotheses on the evolution of separate sexes in flowers (dicliny) have been proposed, both anchored in Lloyd’s views and Darwin’s legacy, with selfing avoidance and optimal limited resource allocation. Intermediate sex separation, in which sex morphs have different combinations of unisexual and hermaphrodite flowers, has been crucial for testing these hypotheses through comparative analyses of optimal conditions in suggested transitions. Again, cases in which floral unisexuality does not lead to sex separation have been studied much less than dioecious plants, at both the microevolutionary and macroevolutionary levels. It is surprising that the increasing availability of plant phylogenies and powerful methods for testing evolutionary transitions and correlations have not led to more studies, even though the frequency of monoecy is probably highest among diclinous species (those with unisexual flowers in any distribution among plants within a population [6]).
The study by Torices et al. [7] aims to fill this gap, offering a different perspective to that provided by Diggle & Miller [8] on the evolution of monoecious conditions. The authors use heads of a number of species of the sunflower family (Asteraceae) to test specifically the effect of resource limitation on the expression of sexual morphs within the head. They make use of the very particular and constant architecture of inflorescences in these species (the flower head or “capitulum”) and the diversity of sexual conditions (hermaphrodite, gynomonoecious, monoecious) and their spatial pattern within the flower head in this plant family to develop an elegant means of testing this hypothesis. Their results are consistent with their expectations on the effect of resource limitation on the head, as determined by patterns of fruit size within the head, assuming that female fecundity is more strongly limited by resource availability than male function.
The authors took on a huge challenge in choosing to study the largest plant family (about 25 thousand species). Their sample was limited to only about a hundred species, but species selection was very careful, to ensure that the range of sex conditions and the available phylogenetic information were adequately represented. The analytical methods are robust and cast no doubt on the reported results. However, I can’t help but wonder what would happen if the antiselfing hypothesis was tested simultaneously. This would require self-incompatibility (SI) data for the species sample, as the presence of SI is usually invoked as a powerful antiselfing mechanism, rendering the unisexuality of flowers unnecessary. However, SI is variable and frequently lost in the sunflower family [9]. I also wonder to what extent the very specific architecture of flower heads imposes an idiosyncratic resource distribution that may have fixed these sexual systems in species and lineages of the family. Although not approached in this study, intraspecific variation seems to be low. It would be very interesting to use similar approaches in other plant groups in which inflorescence architecture is lax and resource distribution may differ. A whole-plant approach might be required, rather than investigations of single inflorescences as in this study. This study has no flaws, but instead paves the way for further testing of a long-standing dual hypothesis, probably with different outcomes in different ecological and evolutionary settings. In the end, sex is not only in the head.
References
[1] Darwin, C. (1877). The different forms of flowers on plants of the same species. John Murray.
[2] Barrett, S. C. H., and Harder, L. D. (2006). David G. Lloyd and the evolution of floral biology: from natural history to strategic analysis. In L.D. Harder, L. D., and Barrett, S. C. H. (eds) Ecology and Evolution of Flowers. OUP, Oxford. Pp 1-21.
[3] Geber, M. A., Dawson, T. E., and Delph, L. F. (eds) (1999). Gender and sexual dimorphism in flowering plants. Springer, Berlin.
[4] Charlesworth, D. (1999). Theories of the evolution of dioecy. In Geber, M. A., Dawson T. E. and Delph L. F. (eds) (1999). Gender and sexual dimorphism in flowering plants. Springer, Berlin. Pp. 33-60.
[5] Friedman, J., and Barrett, S. C. (2008). A phylogenetic analysis of the evolution of wind pollination in the angiosperms. International Journal of Plant Sciences, 169(1), 49-58. doi: 10.1086/523365
[6] Renner, S. S. (2014). The relative and absolute frequencies of angiosperm sexual systems: dioecy, monoecy, gynodioecy, and an updated online database. American Journal of botany, 101(10), 1588-1596. doi: 10.3732/ajb.1400196
[7] Torices, R., Afonso, A., Anderberg, A. A., Gómez, J. M., and Méndez, M. (2019). Architectural traits constrain the evolution of unisexual flowers and sexual segregation within inflorescences: an interspecific approach. bioRxiv, 356147, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/356147
[8] Diggle, P. K., and Miller, J. S. (2013). Developmental plasticity, genetic assimilation, and the evolutionary diversification of sexual expression in Solanum. American journal of botany, 100(6), 1050-1060. doi: 10.3732/ajb.1200647
[9] Ferrer, M. M., and Good‐Avila, S. V. (2007). Macrophylogenetic analyses of the gain and loss of self‐incompatibility in the Asteraceae. New Phytologist, 173(2), 401-414. doi: 10.1111/j.1469-8137.2006.01905.x
| Architectural traits constrain the evolution of unisexual flowers and sexual segregation within inflorescences: an interspecific approach | Rubén Torices, Ana Afonso, Arne A. Anderberg, José M. Gómez and Marcos Méndez | <p>Male and female unisexual flowers have repeatedly evolved from the ancestral bisexual flowers in different lineages of flowering plants. This sex specialization in different flowers often occurs within inflorescences. We hypothesize that inflor... |  | Evolutionary Ecology, Morphological Evolution, Phenotypic Plasticity, Reproduction and Sex, Sexual Selection | Juan Arroyo | Jana Vamosi, Marcial Escudero, Anonymous | 2018-06-27 10:49:52 | View |







