
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields | Recommender▲ | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
17 Nov 2017

ABC random forests for Bayesian parameter inferenceMachine learning methods are useful for Approximate Bayesian Computation in evolution and ecologyRecommended by Michael Blum based on reviews by Dennis Prangle and Michael BlumIt is my pleasure to recommend the paper by Raynal et al. [1] about using random forest for parameter inference. There are two reviews about the paper, one review written by Dennis Prangle and another review written by myself. Both reviews were positive and included comments that have been addressed in the current version of the preprint. The paper nicely shows that modern machine learning approaches are useful for Approximate Bayesian Computation (ABC) and more generally for simulation-driven parameter inference in ecology and evolution. The authors propose to consider the random forest approach, proposed by Meinshausen [2] to perform quantile regression. The numerical implementation of ABC with random forest, available in the abcrf package, is based on the RANGER R package that provides a fast implementation of random forest for high-dimensional data. According to my reading of the manuscript, there are 3 main advantages when using random forest (RF) for parameter inference with ABC. The first advantage is that RF can handle many summary statistics and that dimension reduction is not needed when using RF. The second advantage is very nicely displayed in Figure 5, which shows the main result of the paper. If correct, 95% posterior credibility intervals (C.I.) should contain 95% of the parameter values used in simulations. Figure 5 shows that posterior C.I. obtained with rejection are too large compared to other methods. By contrast, C.I. obtained with regression methods have been shrunken. However, the shrinkage can be excessive for the smallest tolerance rates, with coverage values that can be equal to 85% instead of the expected 95% value. The attractive property of RF is that C.I. have been shrunken but the coverage is of 100% resulting in a conservative decision about parameter values. The last advantage is that no hyperparameter should be chosen. It is a parameter free approach, which is desirable because of the potential difficulty of choosing an appropriate acceptance rate. The main drawback of the proposed approach concerns joint parameter inference. There are many settings where the joint parameter distribution is of interest and the proposed RF approach cannot handle that. In population genetics for example, estimation of the severity and of the duration of the bottleneck should be estimated jointly because of identifiability issues. The challenge of performing joint parameter inference with RF might constitute a useful research perspective. References [1] Raynal L, Marin J-M, Pudlo P, Ribatet M, Robert CP, Estoup A. 2017. ABC random forests for Bayesian parameter inference. arXiv 1605.05537v4, https://arxiv.org/pdf/1605.05537 | ABC random forests for Bayesian parameter inference | Louis Raynal, Jean-Michel Marin, Pierre Pudlo, Mathieu Ribatet, Christian P. Robert, Arnaud Estoup | This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (http:// dx.doi.org/ 10.24072/ pci.evolbiol.100036). Approximate Bayesian computation (ABC) has grown into a standard methodology that manages Bayesian infer... | | Bioinformatics & Computational Biology, Evolutionary Applications, Other, Population Genetics / Genomics | Michael Blum | 2017-07-06 07:42:00 | ||
26 Sep 2017

Lacking conservation genomics in the giant Galápagos tortoiseA genomic perspective is needed for the re-evaluation of species boundaries, evolutionary trajectories and conservation strategies for the Galápagos giant tortoisesRecommended by Michael C. Fontaine based on reviews by 4 anonymous reviewersGenome-wide data obtained from even a small number of individuals can provide unprecedented levels of detail about the evolutionary history of populations and species [1], determinants of genetic diversity [2], species boundaries and the process of speciation itself [3]. Loire and Galtier [4] present a clear example, using the emblematic Galápagos giant tortoise (Chelonoidis nigra), of how multi-species comparative population genomic approaches can provide valuable insights about population structure and species delimitation even when sample sizes are limited but the number of loci is large and distributed across the genome. Galápagos giant tortoises are endemic to the Galápagos Islands and are currently recognized as an endangered, multi-species complex including both extant and extinct taxa. Taxonomic definitions are based on morphology, geographic isolation and population genetic evidence based on short DNA sequences of the mitochondrial genome (mtDNA) and/or a dozen or so nuclear microsatellite loci [5-8]. The species complex enjoys maximal protection. Population recoveries have been quite successful and spectacular conservation programs based on mitochondrial genes and microsatellites are ongoing. This includes for example individual translocations, breeding program, “hybrid” sterilization or removal, and resurrection of extinct lineages). In 2013, Loire et al. [9] provided the first population genomic analyses based on genome scale data (~1000 coding loci derived from blood-transcriptomes) from five individuals, encompassing three putative “species”: Chelonnoidis becki, C. porteri and C. vandenburghi. Their results raised doubts about the validity/accuracy of the currently accepted designations of “genetic distinctiveness”. However, the implications for conservation and management have remained unnoticed. In 2017, Loire and Galtier [4] have re-appraised this issue using an original multi-species comparative population genomic analysis of their previous data set [9]. Based on a comparison of 53 animal species, they show that both the level of genome-wide neutral diversity (πS) and level of population structure estimated using the inbreeding coefficient (F) are much lower than would be expected from a sample covering multiple species. The observed values are more comparable to those typically reported at the “among population” level within a single species such as human (Homo sapiens). The authors go to great length to assess the sensitivity of their method to detect population structure (or lack thereof) and show that their results are robust to potential issues, such as contamination and sequencing errors that can occur with Next Generation Sequencing techniques; and biases related to the small sample size and sub-sampling of individuals. They conclude that published mtDNA and microsatellite-based assessment of population structure and species designations may be biased towards over-splitting. This manuscript is a very good read as it shows the potential of the now relatively affordable genome-wide data for helping to both resolve and clarify population and species boundaries, illuminate demographic trends, evolutionary trajectories of isolated groups, patterns of connectivity among them, and test for evidence of local adaptation and even reproductive isolation. The comprehensive information provided by genome-wide data can critically inform and assist the development of the best strategies to preserve endangered populations and species. Loire and Galtier [4] make a strong case for applying genomic data to the Galápagos giant tortoises, which is likely to redirect conservation efforts more effectively and at lower cost. The case of the Galápagos giant tortoises is certainly a very emblematic example, which will find an echo in many other endangered species conservation programs. References [1] Li H and Durbin R. 2011. Inference of human population history from individual whole-genome sequences. Nature, 475: 493–496. doi: 10.1038/nature10231 [2] Romiguier J, Gayral P, Ballenghien M, Bernard A, Cahais V, Chenuil A, Chiari Y, Dernat R, Duret L, Faivre N, Loire E, Lourenco JM, Nabholz B, Roux C, Tsagkogeorga G, Weber AA-T, Weinert LA, Belkhir K, Bierne N, Glémin S and Galtier N. 2014. Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature, 515: 261–263. doi: 10.1038/nature13685 [3] Roux C, Fraïsse C, Romiguier J, Anciaux Y, Galtier N and Bierne N. 2016. Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS Biology, 14: e2000234. doi: 10.1371/journal.pbio.2000234 [4] Loire E and Galtier N. 2017. Lacking conservation genomics in the giant Galápagos tortoise. bioRxiv 101980, ver. 4 of September 26, 2017. doi: 10.1101/101980 [5] Beheregaray LB, Ciofi C, Caccone A, Gibbs JP and Powell JR. 2003. Genetic divergence, phylogeography and conservation units of giant tortoises from Santa Cruz and Pinzón, Galápagos Islands. Conservation Genetics, 4: 31–46. doi: 10.1023/A:1021864214375 [6] Ciofi C, Milinkovitch MC, Gibbs JP, Caccone A and Powell JR. 2002. Microsatellite analysis of genetic divergence among populations of giant Galápagos tortoises. Molecular Ecology, 11: 2265–2283. doi: 10.1046/j.1365-294X.2002.01617.x [7] Garrick RC, Kajdacsi B, Russello MA, Benavides E, Hyseni C, Gibbs JP, Tapia W and Caccone A. 2015. Naturally rare versus newly rare: demographic inferences on two timescales inform conservation of Galápagos giant tortoises. Ecology and Evolution, 5: 676–694. doi: 10.1002/ece3.1388 [8] Poulakakis N, Edwards DL, Chiari Y, Garrick RC, Russello MA, Benavides E, Watkins-Colwell GJ, Glaberman S, Tapia W, Gibbs JP, Cayot LJ and Caccone A. 2015. Description of a new Galápagos giant tortoise species (Chelonoidis; Testudines: Testudinidae) from Cerro Fatal on Santa Cruz island. PLoS ONE, 10: e0138779. doi: 10.1371/journal.pone.0138779 [9] Loire E, Chiari Y, Bernard A, Cahais V, Romiguier J, Nabholz B, Lourenço JM and Galtier N. 2013. Population genomics of the endangered giant Galápagos tortoise. Genome Biology, 14: R136. doi: 10.1186/gb-2013-14-12-r136 | Lacking conservation genomics in the giant Galápagos tortoise | Etienne Loire, Nicolas Galtier | <p>Conservation policy in the giant Galápagos tortoise, an iconic endangered animal, has been assisted by genetic markers for ~15 years: a dozen loci have been used to delineate thirteen (sub)species, between which hybridization is prevented. Here... | | Evolutionary Applications, Population Genetics / Genomics, Speciation, Systematics / Taxonomy | Michael C. Fontaine | 2017-01-21 15:34:00 | ||
04 Jul 2022
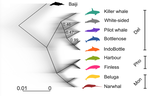
A genomic assessment of the marine-speciation paradox within the toothed whale superfamily DelphinoideaReticulated evolution marks the rapid diversification of the DelphinoidaeRecommended by Michael C. Fontaine based on reviews by Christelle Fraïsse, Simon Henry Martin, Andrew Foote and 2 anonymous reviewersHistorically neglected or considered a rare aberration in animals under the biological species concept, interspecific hybridisation has by now been recognised to be taxonomically widespread, particularly in rapidly diversifying groups (Dagilis et al. 2021; Edelman & Mallet 2021; Mallet et al. 2016; Seehausen 2004). Yet the prevalence of introgressive hybridizations, its evolutionary significance, and its impact on species diversification remain a hot topic of research in evolutionary biology. The rapid increase in genomic resources now available for non-model species has significantly contributed to the detection of introgressive hybridization across taxa showing that reticulated evolution is far more common in the animal kingdom than historically considered. Yet, detecting it, quantifying its magnitude, and assessing its evolutionary significance remains a challenging endeavour with constantly evolving methodologies to better explore and exploit genomic data (Blair & Ané 2020; Degnan & Rosenberg 2009; Edelman & Mallet 2021; Hibbins & Hahn 2022). In the marine realm, the dearth of geographic barriers and the large dispersal abilities of pelagic species like cetaceans have raised the questions of how populations and species can diverge and adapt to distinct ecological conditions in face of potentially large gene-flow, the so-called marine speciation paradox (Bierne et al. 2003). Contemporaneous hybridization among cetacean species has been widely documented in nature despite large phenotypic differences (Crossman et al. 2016). The historical prevalence of reticulated evolution, its evolutionary significance, and how it might have impacted the evolutionary history and diversification of the cetaceans have however remained elusive so far. Recent phylogenomic studies suggested that introgression has been prevalent in cetacean evolutionary history with instances reported among baleen whales (mysticetes) (Árnason et al. 2018) and among toothed whales (odontocetes), especially in the rapidly diversifying dolphins family of the Delphininae (Guo et al. 2021; Moura et al. 2020). Analysing publicly available whole-genome data from nine cetacean species across three families of Delphinoidae – dolphins, porpoises, and monondontidae – using phylogenomics and demo-genetics approaches, Westbury and colleagues (2022) take a step further in documenting that evolution among these species has been far from a simple bifurcating tree. Instead, their study describes widespread occurrences of introgression among Delphinoidae, drawing a complex picture of reticulated evolutionary history. After describing major topology discordance in phylogenetic gene trees along the genome, the authors use a panel of approaches to disentangle introgression from incomplete lineage sorting (ILS), the two most common causes of tree topology discordances (Hibbins & Hahn 2022). Applying popular tests that separate introgression from ILS, such as the Patterson’s D (a.k.a. ABBA-BABA) test (Durand et al. 2011; Green et al. 2010), QuIBL (Edelman et al. 2019), and D-FOIL (Pease & Hahn 2015), the authors report that signals of introgression are present in the genomes of most (if not all) the cetacean species included in their study. However, this picture needs to be nuanced. Most introgression signals seem to echo old introgression events that occurred primarily among ancestors. This is suggested by the differential signals of topology discordance along the genome when considering sliding windows along the genome of varying sizes (50kb, 100kb, and 1Mb), and by patterns of excess derived allele sharing along branches of the species tree, as captured by the f-branch test (Malinsky et al. 2021; Malinsky et al. 2018). The authors further investigated the dynamic of cessation of gene flow (and/or ILS) between species using the F1 hybrid PSMC (or hPSMC) approach (Cahill et al. 2016). By estimating the cross-coalescent rates (CRR) between species pairs with time in light of previously estimated species divergence times (McGowen et al. 2020), the authors report that gene flow (and/or ILS) most likely has stopped by now among most species, but it may have lasted for more than half of the time since the species split from each other. According to the author, this result may reflect the slow process by which reproductive isolation would have evolved between cetacean lineages, and that species isolation was marked by significant introgression events. Now, while the present study provides a good overview of how complex is the reticulated evolutionary history of the Delphinoidae, getting a complete picture will require overcoming a few important limitations. The first ones are methodological and related to the phylogenomic analyses. Given the sampling design with one diploid genome per species, the authors could not phase the data into the parental haplotypes, but instead relied on a majority consensus creating mosaic haploidized genomes representing a mixture between the two parental copies. Moreover, by using large genomic windows (≥50kb) that likely span multiple independent loci, phylogenetic analyses in windows encompassed distinct phylogenetic signals, potentially leading to bias and inaccuracy in the inferences. Thawornwattana et al (2018) previously showed that this “concatenation approach” could significantly impact phylogenetic inferences. They proposed instead to use loci small enough to minimise the risk of intra-locus recombination and to consider them in blocks of non-recombining loci along the genome in order to conduct phylogenetic analysed, ideally under the multi-species coalescent (MSC) that can account for ILS (e.g. BPP; Flouri et al. 2018; Jiao et al. 2020; Yang 2015). Such an approach applied to the diversification of the Delphinidae may reveal substantial changes compared to the currently admitted species tree. Inaccuracy in the species tree estimation may have a major impact on the introgression analyses conducted in this study since the species tree and branching order must be supplied in the introgression analyses to properly disentangle introgression from ILS. Here, the authors rely on the tree topology that was previously estimated in McGowen et al. (2020), which they also recovered using their consensus estimation from ASTRAL-III (Zhang et al. 2018). While the methodologies accounted to a certain extent for ILS, albeit with potential bias induced by the concatenation approach, they ignore the presumably large amount of introgression among species during the diversification process. Estimating species branching order while ignoring introgression can lead to major bias in the phylogenetic inference and can lead to incorrect topologies. Even if these MSC-based methods account for ILS, inferences can become very inaccurate or even break down as gene flow increases (see for ex. Jiao et al. 2020; Müller et al. in press; Solís-Lemus et al. 2016). Dedicated approaches have been developed to model explicitly introgression together with ILS to estimate phylogenetic networks (Blair & Ané 2020; Rabier et al. 2021) or in isolation-with-migration model (Müller et al. in press; Wang et al. 2020). Future studies revisiting the reticulated evolutionary history of the Delphinoidae with such approaches may not only precise the species branching order, but also deliver a finer view of the magnitude and prevalence of introgression during the evolutionary history of these species. A final part of Westbury et al. (2022)'s study set out to test whether historical periods of low abundance could have facilitated hybridization among Delphinoidae species. During these periods of low abundance, species may encounter only a limited number of conspecifics and may consider individuals from other species as suitable mating partners, leading to hybridisation (Crossman et al. 2016; Edwards et al. 2011; Westbury et al. 2019). The authors tested this hypothesis by considering genome-wide genetic diversity (or heterozygosity) as a proxy of historical effective population size (Ne), itself as a proxy of the evolution of census size with time. They also try to link historical Ne variation (from PSMC, Li & Durbin 2011) with their estimated time to cessation of gene flow or ILS (from the CRR of hPSMC). However, no straightforward relationship was found between the genetic diversity and the propensity of species to hybridize, nor was there any clear link between Ne variation through time and the cessation of gene flow or ILS. Such a lack of relationship may not come as a surprise, since the determinants of genome-wide genetic diversity and its variation through evolutionary time-scale are far more diverse and complex than just a direct link with hybridization, introgression, or even with the census population size. In fact, genetic diversity results from the balance between all the evolutionary processes at play in the species' evolutionary history (see the review of Ellegren & Galtier 2016). Other important factors can strongly impact genetic diversity, including demography and structure, but also linked selection (Boitard et al. 2022; Buffalo 2021; Ellegren & Galtier 2016). All in all, Westbury and coll. (2022) present here an interesting study providing an additional step towards resolving and understanding the complex evolutionary history of the Delphinoidae, and shedding light on the importance of introgression during the diversification of these cetacean species. Prospective work improving upon the taxonomic sampling, with additional genomic data for each species, considered with dedicated approaches tailored at estimating species tree or network while accounting for ILS and introgression will be key for refining the picture depicted in this study. Down the road, altogether these studies will contribute to assessing the evolutionary significance of introgression on the diversification of Delphinoides, and more generally on the diversification of cetacean species, which still remain an open and exciting perspective. References Árnason Ú, Lammers F, Kumar V, Nilsson MA, Janke A (2018) Whole-genome sequencing of the blue whale and other rorquals finds signatures for introgressive gene flow. Science Advances 4, eaap9873. https://doi.org/10.1126/sciadv.aap9873 Bierne N, Bonhomme F, David P (2003) Habitat preference and the marine-speciation paradox. Proceedings of the Royal Society of London. Series B: Biological Sciences 270, 1399-1406. https://doi.org/10.1098/rspb.2003.2404 Blair C, Ané C (2020) Phylogenetic Trees and Networks Can Serve as Powerful and Complementary Approaches for Analysis of Genomic Data. Systematic Biology 69, 593-601. https://doi.org/10.1093/sysbio/syz056 Boitard S, Arredondo A, Chikhi L, Mazet O (2022) Heterogeneity in effective size across the genome: effects on the inverse instantaneous coalescence rate (IICR) and implications for demographic inference under linked selection. Genetics 220, iyac008. https://doi.org/10.1093/genetics/iyac008 Buffalo V (2021) Quantifying the relationship between genetic diversity and population size suggests natural selection cannot explain Lewontin's Paradox. e-Life 10, e67509. https://doi.org/10.7554/eLife.67509 Cahill JA, Soares AE, Green RE, Shapiro B (2016) Inferring species divergence times using pairwise sequential Markovian coalescent modelling and low-coverage genomic data. Philos Trans R Soc Lond B Biol Sci 371, 20150138. https://doi.org/10.1098/rstb.2015.0138 Crossman CA, Taylor EB, Barrett‐Lennard LG (2016) Hybridization in the Cetacea: widespread occurrence and associated morphological, behavioral, and ecological factors. Ecology and Evolution 6, 1293-1303. https://doi.org/10.1002/ece3.1913 Dagilis AJ, Peede D, Coughlan JM, Jofre GI, D’Agostino ERR, Mavengere H, Tate AD, Matute DR (2021) 15 years of introgression studies: quantifying gene flow across Eukaryotes. biorXiv, 2021.1106.1115.448399. https://doi.org/10.1101/2021.06.15.448399 Degnan JH, Rosenberg NA (2009) Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol Evol 24, 332-340. https://doi.org/10.1016/j.tree.2009.01.009 Durand EY, Patterson N, Reich D, Slatkin M (2011) Testing for ancient admixture between closely related populations. Mol Biol Evol 28, 2239-2252. https://doi.org/10.1093/molbev/msr048 Edelman NB, Frandsen PB, Miyagi M, Clavijo B, Davey J, Dikow RB, Garcia-Accinelli G, Van Belleghem SM, Patterson N, Neafsey DE, Challis R, Kumar S, Moreira GRP, Salazar C, Chouteau M, Counterman BA, Papa R, Blaxter M, Reed RD, Dasmahapatra KK, Kronforst M, Joron M, Jiggins CD, McMillan WO, Di Palma F, Blumberg AJ, Wakeley J, Jaffe D, Mallet J (2019) Genomic architecture and introgression shape a butterfly radiation. Science 366, 594-599. https://doi.org/10.1126/science.aaw2090 Edelman NB, Mallet J (2021) Prevalence and Adaptive Impact of Introgression. Annual Review of Genetics 55, 265-283. https://doi.org/10.1146/annurev-genet-021821-020805 Edwards CJ, Suchard MA, Lemey P, Welch JJ, Barnes I, Fulton TL, Barnett R, O'Connell TC, Coxon P, Monaghan N, Valdiosera CE, Lorenzen ED, Willerslev E, Baryshnikov GF, Rambaut A, Thomas MG, Bradley DG, Shapiro B (2011) Ancient hybridization and an Irish origin for the modern polar bear matriline. Curr Biol 21, 1251-1258. https://doi.org/10.1016/j.cub.2011.05.058 Ellegren H, Galtier N (2016) Determinants of genetic diversity. Nat Rev Genet 17, 422-433. https://doi.org/10.1038/nrg.2016.58 Flouri T, Jiao X, Rannala B, Yang Z (2018) Species Tree Inference with BPP Using Genomic Sequences and the Multispecies Coalescent. Mol Biol Evol 35, 2585-2593. https://doi.org/10.1093/molbev/msy147 Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, Hansen NF, Durand EY, Malaspinas AS, Jensen JD, Marques-Bonet T, Alkan C, Prufer K, Meyer M, Burbano HA, Good JM, Schultz R, Aximu-Petri A, Butthof A, Hober B, Hoffner B, Siegemund M, Weihmann A, Nusbaum C, Lander ES, Russ C, Novod N, Affourtit J, Egholm M, Verna C, Rudan P, Brajkovic D, Kucan Z, Gusic I, Doronichev VB, Golovanova LV, Lalueza-Fox C, de la Rasilla M, Fortea J, Rosas A, Schmitz RW, Johnson PLF, Eichler EE, Falush D, Birney E, Mullikin JC, Slatkin M, Nielsen R, Kelso J, Lachmann M, Reich D, Paabo S (2010) A draft sequence of the Neandertal genome. Science 328, 710-722. https://doi.org/10.1126/science.1188021 Guo W, Sun D, Cao Y, Xiao L, Huang X, Ren W, Xu S, Yang G (2021) Extensive Interspecific Gene Flow Shaped Complex Evolutionary History and Underestimated Species Diversity in Rapidly Radiated Dolphins. Journal of Mammalian Evolution 29, 353-367. https://doi.org/10.1007/s10914-021-09581-6 Hibbins MS, Hahn MW (2022) Phylogenomic approaches to detecting and characterizing introgression. Genetics 220, iyab173. https://doi.org/10.1093/genetics/iyab173 Jiao X, Flouri T, Rannala B, Yang Z (2020) The Impact of Cross-Species Gene Flow on Species Tree Estimation. Syst Biol 69, 830-847. https://doi.org/10.1093/sysbio/syaa001 Li H, Durbin R (2011) Inference of human population history from individual whole-genome sequences. Nature 475, 493-496. https://doi.org/10.1038/nature10231 Malinsky M, Matschiner M, Svardal H (2021) Dsuite - Fast D-statistics and related admixture evidence from VCF files. Mol Ecol Resour 21, 584-595. https://doi.org/10.1111/1755-0998.13265 Malinsky M, Svardal H, Tyers AM, Miska EA, Genner MJ, Turner GF, Durbin R (2018) Whole-genome sequences of Malawi cichlids reveal multiple radiations interconnected by gene flow. Nature Ecology & Evolution 2, 1940-1955. https://doi.org/10.1038/s41559-018-0717-x Mallet J, Besansky N, Hahn MW (2016) How reticulated are species? Bioessays 38, 140-149. https://doi.org/10.1002/bies.201500149 McGowen MR, Tsagkogeorga G, Alvarez-Carretero S, Dos Reis M, Struebig M, Deaville R, Jepson PD, Jarman S, Polanowski A, Morin PA, Rossiter SJ (2020) Phylogenomic Resolution of the Cetacean Tree of Life Using Target Sequence Capture. Syst Biol 69, 479-501. https://doi.org/10.1093/sysbio/syz068 Moura AE, Shreves K, Pilot M, Andrews KR, Moore DM, Kishida T, Möller L, Natoli A, Gaspari S, McGowen M, Chen I, Gray H, Gore M, Culloch RM, Kiani MS, Willson MS, Bulushi A, Collins T, Baldwin R, Willson A, Minton G, Ponnampalam L, Hoelzel AR (2020) Phylogenomics of the genus Tursiops and closely related Delphininae reveals extensive reticulation among lineages and provides inference about eco-evolutionary drivers. Molecular Phylogenetics and Evolution 146,107047. https://doi.org/10.1016/j.ympev.2020.106756 Müller NF, Ogilvie HA, Zhang C, Fontaine MC, Amaya-Romero JE, Drummond AJ, Stadler T (in press) Joint inference of species histories and gene flow. Syst Biol. Pease JB, Hahn MW (2015) Detection and Polarization of Introgression in a Five-Taxon Phylogeny. Syst Biol 64, 651-662. https://doi.org/10.1093/sysbio/syv023 Rabier CE, Berry V, Stoltz M, Santos JD, Wang W, Glaszmann JC, Pardi F, Scornavacca C (2021) On the inference of complex phylogenetic networks by Markov Chain Monte-Carlo. PLoS Comput Biol 17, e1008380. https://doi.org/10.1371/journal.pcbi.1008380 Seehausen O (2004) Hybridization and adaptive radiation. Trends Ecol Evol 19, 198-207. https://doi.org/10.1016/j.tree.2004.01.003 Solís-Lemus C, Yang M, Ané C (2016) Inconsistency of Species Tree Methods under Gene Flow. Syst Biol 65, 843-851. https://doi.org/10.1093/sysbio/syw030 Thawornwattana Y, Dalquen D, Yang Z, Tamura K (2018) Coalescent Analysis of Phylogenomic Data Confidently Resolves the Species Relationships in the Anopheles gambiae Species Complex. Molecular Biology and Evolution 35, 2512-2527. https://doi.org/10.1093/molbev/msy158 Wang K, Mathieson I, O’Connell J, Schiffels S (2020) Tracking human population structure through time from whole genome sequences. PLOS Genetics 16, e1008552. https://doi.org/10.1371/journal.pgen.1008552 Westbury MV, Cabrera AA, Rey-Iglesia A, Cahsan BD, Duchêne DA, Hartmann S, Lorenzen ED (2022) A genomic assessment of the marine-speciation paradox within the toothed whale superfamily Delphinoidea. bioRxiv, 2020.10.23.352286, ver. 7 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.10.23.352286 Westbury MV, Petersen B, Lorenzen ED (2019) Genomic analyses reveal an absence of contemporary introgressive admixture between fin whales and blue whales, despite known hybrids. PLoS ONE 14, e0222004. https://doi.org/10.1371/journal.pone.0222004 Yang Z (2015) The BPP program for species tree estimation and species delimitation. Current Zoology 61, 854-865. https://doi.org/10.1093/czoolo/61.5.854 Zhang C, Rabiee M, Sayyari E, Mirarab S (2018) ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19, 153. https://doi.org/10.1186/s12859-018-2129-y | A genomic assessment of the marine-speciation paradox within the toothed whale superfamily Delphinoidea | Michael V Westbury, Andrea A Cabrera, Alba Rey-Iglesia, Binia De Cahsan, David A. Duchêne, Stefanie Hartmann, Eline D Lorenzen | <p>The importance of post-divergence gene flow in speciation has been documented across a range of taxa in recent years, and may have been especially widespread in highly mobile, wide-ranging marine species, such as cetaceans. Here, we studied ind... | | Evolutionary Dynamics, Hybridization / Introgression, Molecular Evolution, Phylogenetics / Phylogenomics, Speciation | Michael C. Fontaine | 2020-10-25 08:55:50 | ||
16 Dec 2016

POSTPRINT
Evolutionary robotics simulations help explain why reciprocity is rare in nature.Simulated robots and the evolution of reciprocityRecommended by Michael D Greenfield and Joël Meunier
Of the various forms of cooperative and altruistic behavior, reciprocity remains the most contentious. Humans certainly exhibit reciprocity – under certain circumstances – and various non-human animals behave in ways suggesting that they do as well. Thus, evolutionary biologists have sought to explain why non-relatives might engage in altruistic transactions when a substantial delay occurs between helping and compensation; i.e. an individual may be a donor today and a beneficiary tomorrow, or vice-versa. This quest, aided by game theory and computer modeling late in the past century, identified some strategies for reciprocal behavior that could work – in theory. But when biologists looked for confirmation of these strategies in animals they found little evidence that stood up to rigorous testing. In a recent paper André and Nolfi [1] offer a compelling reason for this observed rarity of reciprocity: Reciprocal behavior that animals might exhibit is a bit more complex than any of the game theoretic strategies, and even the simplest forms of realistic behavior would entail several nearly simultaneous mutations, an unlikely occurrence. André and Nolfi [1] relied on neural networks to test actors, robots that could evolve helping and reciprocal behavior from a basal level of selfishness. In an extensive series of simulations, they found that reciprocal behavior did not take hold in a population, largely because the various intermediates to full reciprocity were eliminated before the subsequent mutations occurred. The findings are satisfying given our current knowledge of animal behavior, but questions remain. Notably, how does one account for those rare cases in which reciprocity does meet all the criteria? The authors suggest some possibilities, but an analysis will await their next study. Reference [1] André J-B, Nolfi S. 2016. Evolutionary robotics simulations help explain why reciprocity is rare in nature. Scientific Reports 6:32785. doi: 10.1038/srep32785 | Evolutionary robotics simulations help explain why reciprocity is rare in nature. | André J-B, Nolfi S | The relative rarity of reciprocity in nature, contrary to theoretical predictions that it should be widespread, is currently one of the major puzzles in social evolution theory. Here we use evolutionary robotics to solve this puzzle. We show tha... | | Behavior & Social Evolution, Evolutionary Theory | Michael D Greenfield | 2016-12-16 18:08:31 | ||
18 Nov 2020
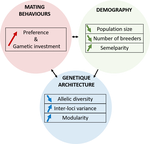
A demogenetic agent based model for the evolution of traits and genome architecture under sexual selectionSexual selection goes dynamicRecommended by Michael D Greenfield based on reviews by Frédéric Guillaume and 1 anonymous reviewer150 years after Darwin published ‘Descent of man and selection in relation to sex’ (Darwin, 1871), the evolutionary mechanism that he laid out in his treatise continues to fascinate us. Sexual selection is responsible for some of the most spectacular traits among animals, and plants, and it appeals to our interest in all things reproductive and sexual (Bell, 1982). In addition, sexual selection poses some of the more intractable problems in evolutionary biology: Its realm encompasses traits that are subject to markedly different selection pressures, particularly when distinct, yet associated, traits tend to be associated with males, e.g. courtship signals, and with females, e.g. preferences (cf. Ah-King & Ahnesjo, 2013). While separate, such traits cannot evolve independently of each other (Arnqvist & Rowe, 2005), and complex feedback loops and correlations between them are predicted (Greenfield et al., 2014). Traditionally, sexual selection has been modelled under simplifying assumptions, and quantitative genetic approaches that avoided evolutionary dynamics have prevailed. New computing methods may be able to free the field from these constraints, and a trio of theoreticians (Chevalier, De Coligny & Labonne 2020) describe here a novel application of a ‘demo-genetic agent (or individual) based model’, a mouthful hereafter termed DG-ABM, for arriving at a holistic picture of the sexual selection trajectory. The application is built on the premise that traits, e.g. courtship, preference, gamete investment, competitiveness for mates, can influence the genetic architecture, e.g. correlations, of those traits. In turn, the genetic architecture can influence the expression and evolvability of the traits. Much of this influence occurs via demographic features, i.e. social environment, generated by behavioral interactions during sexual advertisement, courtship, mate guarding, parental care, post-mating dispersal, etc. References Ah-King, M. and Ahnesjo, I. 2013. The ‘sex role’ concept: An overview and evaluation Evolutionary Biology, 40, 461-470. doi: https://doi.org/10.1007/s11692-013-9226-7 | A demogenetic agent based model for the evolution of traits and genome architecture under sexual selection | Louise Chevalier, François de Coligny, Jacques Labonne | <p>Sexual selection has long been known to favor the evolution of mating behaviors such as mate preference and competitiveness, and to affect their genetic architecture, for instance by favoring genetic correlation between some traits. Reciprocall... | | Adaptation, Behavior & Social Evolution, Evolutionary Dynamics, Evolutionary Theory, Life History, Population Genetics / Genomics, Sexual Selection | Michael D Greenfield | 2020-04-02 14:44:25 | ||
29 Nov 2023

Does sociality affect evolutionary speed?On the evolutionary implications of being a social animalRecommended by Michael D Greenfield based on reviews by Rafael Lucas Rodriguez and 1 anonymous reviewerWhat does it mean to be highly social? Considering the so-called four ‘pinnacles’ of animal society (Wilson, 1975) – humans, cooperative breeding as found in some non-human mammals and birds, the social insects, and colonial marine invertebrates – having inter-individual relations extending beyond the sexual pair and the parent-offspring interaction is foremost. In many cases being social implies a high local population density, interaction with the same group of individuals over an extended time period, and an overlapping of generations. Additional features of social species may be a wide geographical range, perhaps associated with ecological and behavioral plasticity, the latter often facilitated by cultural transmission of traditions. Narrowing our perspective to the domain of PCI Evolutionary Biology, we might continue our question by asking whether being social predisposes one to a special evolutionary path toward the future. Do social species evolve faster (or slower) than their more solitary relatives such that over time they are more unlike (or similar to) those relatives (anagenesis)? And are evolutionary changes in social species more or less likely to be accompanied by lineage splitting (cladogenesis) and ultimately speciation? The latter question is parallel to one first posed over 40 years ago (West-Eberhard, 1979; Lande, 1981) for sexually selected traits: Do strong mating preferences and conspicuous courtship signals generate speciation via the Fisherian process or ecological divergence? An extensive survey of birds had found little supporting evidence (Price, 1998), but a recent one that focused on plumage complexity in tanagers did reveal a relationship, albeit a weak one (Price-Waldman et al., 2020). Because sexual selection has been viewed as a part of the broader process of social selection (West-Eberhard, 1979), it is thus fitting to extend our surveys to the evolutionary implications of being social. Unlike the inquiry for a sexual selection - evolutionary change connection, a social behavior counterpart has remained relatively untreated. Diverse logistical problems might account for this oversight. What objective proxies can be used for social behavior, and for the rate of evolutionary change within a lineage? How many empirical studies have generated data from which appropriate proxies could be extracted? More intractable is the conundrum arising from the connectedness between socially- and sexually-selected traits. For example, the elevated population density found in highly social species can greatly increase the mating advantage enjoyed by an attractive male. If anagenesis is detected, did it result from social behavior or sexual selection? And if social behavior leads to a group structure in which male-male competition is reduced, would a modest rate of evolutionary change be support for the sexual selection - evolutionary speed connection or evidence opposing the sociality - evolution one? Against the above odds, several biologists have begun to explore the notion that social behavior just might favor evolutionary speed in either anagenesis or cladogenesis. In a recent analysis relying on the comparative method, Lluís Socias-Martínez and Louise Rachel Peckre (2023) combed the scientific literature archives and identified those studies with specific data on the relationships between sexual selection or social behavior and evolutionary change, either anagenesis or cladogenesis. The authors were careful to employ fairly conservative criteria for including studies, and the number eventually retained was small. Nonetheless, some patterns emerge: Many more studies report anagenesis than cladogenesis, and many more report correlations with sexually-selected traits than with non-sexual social behavior ones. And, no study indicates a potential effect of social behavior on cladogenesis. Is this latter observation authentic or an artifact of a paucity of data? There are some a priori reasons why cladogenesis may seldom arise. Whereas highly social behavior could lead to fission encompassing mutually isolated population clusters within a species, social behavior may also engender counterbalancing plasticity that allows and even promotes inter-cluster migration and fusion. And briefly – and non-systematically, as the rate of lineage splitting would need to be measured – looking at one of the pinnacles of animal social behavior, the social insects, there is little indication that diversification has been accelerated. There are fewer than 3000 described species of termites, only ca. 16,000 ants, and the vast majority of bees and wasps are solitary. Lluís Socias-Martínez and Louise Rachel Peckre provide us with a very detailed discussion of these and a myriad of other complications. I end with a common refrain, we need more consideration of the authors’ interesting question, and much more data and analysis. One can thank Socias-Martínez and Peckre for pointing us in that direction. References Lande, R. (1981). Models of speciation by sexual selection on polygenic traits. Proc. Natn. Acad. Sci. USA 78, 3721-3725. https://doi.org/10.1073/pnas.78.6.3721 Price, T. (1998). Sexual selection and natural selection in bird speciation. Phil. Trans. Roy. Soc. B, 353, 251-260. https://doi.org/10.1098/rstb.1998.0207 Price‐Waldman, R. M., Shultz, A. J., & Burns, K. J. (2020). Speciation rates are correlated with changes in plumage color complexity in the largest family of songbirds. Evolution, 74(6), 1155–1169. https://doi.org/10.1111/evo.13982 Socias-Martínez and Peckre. (2023). Does sociality affect evolutionary speed? Zenodo, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.5281/zenodo.10086186 West-Eberhard, M. J. (1979). Sexual selection, social competition, and evolution. Proceedings of the American Philosophical Society, 123(4), 222–234. http://www.jstor.org/stable/2828804 Wilson, E. O. (1975). Sociobiology. The New Synthesis. Cambridge, Mass., The Belknap Press of Harvard University | Does sociality affect evolutionary speed? | Lluís Socias-Martínez, Louise Rachel Peckre | <p>An overlooked source of variation in evolvability resides in the social lives of animals. In trying to foster research in this direction, we offer a critical review of previous work on the link between evolutionary speed and sociality. A first ... | | Behavior & Social Evolution, Evolutionary Dynamics, Evolutionary Theory, Genome Evolution, Macroevolution, Molecular Evolution, Population Genetics / Genomics, Sexual Selection, Speciation | Michael D Greenfield | 2023-03-03 00:10:49 | ||
11 Mar 2020
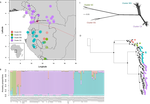
Phylogenomic approaches reveal how a climatic inversion and glacial refugia shape patterns of diversity in an African rain forest tree speciesRemarkable insights into processes shaping African tropical tree diversityRecommended by Michael Pirie based on reviews by Miguel de Navascués, Lars Chatrou and Oscar Vargas
Tropical biodiversity is immense, under enormous threat, and yet still poorly understood. Global climatic breakdown and habitat destruction are impacting on and removing this diversity before we can understand how the biota responds to such changes, or even fully appreciate what we are losing [1]. This is particularly the case for woody shrubs and trees [2] and for the flora of tropical Africa [3]. Helmstetter et al. [4] have taken a significant step to improve our understanding of African tropical tree diversity in the context of past climatic change. They have done so by means of a remarkably in-depth analysis of one species of the tropical plant family Annonaceae: Annickia affinis [5]. A. affinis shows a distribution pattern in Africa found in various plant (but interestingly not animal) groups: a discontinuity between north and south of the equator [6]. There is no obvious physical barrier to cause this discontinuity, but it does correspond with present day distinct northern and southern rainy seasons. Various explanations have been proposed for this discontinuity, set out as hypotheses to be tested in this paper: climatic fluctuations resulting in changes in plant distributions in the Pleistocene, or differences in flowering times or in ecological niche between northerly and southerly populations. These explanations are not mutually exclusive, but they can be tested using phylogenetic inference – if you can sample variable enough sequence data from enough individuals – complemented with analysis of ecological niches and traits. Using targeted sequence capture, the authors amassed a dataset representing 351 nuclear markers for 112 individuals of A. affinis. This dataset is impressive for a number of reasons: First, sampling such a species across such a wide range in tropical Africa presents numerous challenges of itself. Second, the technical achievement of using this still relatively new sequencing technique with a custom set of baits designed specifically for this plant family [7] is also considerable. The result is a volume of data that just a few years ago would not have been feasible to collect, and which now offers the possibility to meaningfully analyse DNA sequence variation within a species across numerous independent loci of the nuclear genome. This is the future of our research field, and the authors have ably demonstrated some of its possibilities. Using this data, they performed on the one hand different population genetic clustering approaches, and on the other, different phylogenetic inference methods. I would draw attention to their use and comparison of coalescence and network-based approaches, which can account for the differences between gene trees that might be expected between populations of a single species. The results revealed four clades and a consistent sequence of divergences between them. The authors inferred past shifts in geographic range (using a continuous state phylogeographic model), depicting a biogeographic scenario involving a dispersal north over the north/south discontinuity; and demographic history, inferring in some (but not all) lineages increases in effective population size around the time of the last glacial maximum, suggestive of expansion from refugia. Using georeferenced specimen data, they compared ecological niches between populations, discovering that overlap was indeed smallest comparing north to south. Just the phenology results were effectively inconclusive: far better data on flowering times is needed than can currently be harvested from digitised herbarium specimens. Overall, the results add to the body of evidence for the impact of Pleistocene climatic changes on population structure, and for niche differences contributing to the present day north/south discontinuity. However, they also paint a complex picture of idiosyncratic lineage-specific responses, even within a single species. With the increasing accessibility of the techniques used here we can look forward to more such detailed analyses of independent clades necessary to test and to expand on these conclusions, better to understand the nature of our tropical plant diversity while there is still opportunity to preserve it for future generations. References [1] Mace, G. M., Gittleman, J. L., and Purvis, A. (2003). Preserving the Tree of Life. Science, 300(5626), 1707–1709. doi: 10.1126/science.1085510 | Phylogenomic approaches reveal how a climatic inversion and glacial refugia shape patterns of diversity in an African rain forest tree species | Andrew J. Helmstetter, Biowa E. N. Amoussou, Kevin Bethune, Narcisse G. Kandem, Romain Glèlè Kakaï, Bonaventure Sonké, Thomas L. P. Couvreur | <p>The world’s second largest expanse of tropical rain forest is in Central Africa and it harbours enormous species diversity. Population genetic studies have consistently revealed significant structure across central African rain forest plants, i... | | Evolutionary Dynamics, Phylogeography & Biogeography | Michael Pirie | 2019-10-29 15:19:36 | ||
12 Nov 2021

How ancient forest fragmentation and riparian connectivity generate high levels of genetic diversity in a micro-endemic Malagasy treeAn ancient age of open-canopy landscapes in northern Madagascar? Evidence from the population genetic structure of a forest treeRecommended by Miguel de Navascués based on reviews by Katharina Budde and Yurena Arjona
We currently live in the Anthropocene, the geological age characterized by a profound impact of human populations in the ecosystems and the environment. While there is little doubt about the action of humans in the shaping of present landscapes, it can be difficult to determine what the state of those landscapes was before humans started to modify them. This is the case of the Madagascar grasslands, whose origins have been debated with arguments proposing them either as anthropogenic, created with the arrival of humans around 2000BP, or as ancient features of the natural landscape with a forest fragmentation process due to environmental changes pre-dating human arrival [e.g. 1,2]. One way to clarify this question is through the genetic study of native species. Population continuity and fragmentation along time shape the structure of the genetic diversity in space. Species living in a uniform continuous habitat are expected to show genetic structuring determined only by geographical distance. Recent changes of the habitat can take many generations to reshape that genetic structure [3]. Thus, we expect genetic structure to reflect ancient features of the landscape. The work by Jordi Salmona and collaborators [4] studies the factors determining the population genetic structure of the Malagasy spiny olive (Noronhia spinifolia). This narrow endemic species is distributed in the discontinuous forest patches of the Loky-Manambato region (northern Madagascar). Jordi Salmona and collaborators genotyped 72 individuals distributed across the species distribution with restriction associated DNA sequencing and organelle microsatellite markers. Then, they studied the population genetic structure of the species. Using isolation-by-resistance models [5], they tested the influence of several landscape features (forest cover, roads, rivers, slope, etc.) on the connectivity between populations. Maternally inherited loci (chloroplast and mitochondria) and bi-parentally inherited loci (nuclear), were analysed separately in an attempt to identify the role of pollen and seed dispersal in the connectivity of populations. Despite the small distribution of the species, Jordi Salmona and collaborators [4] found remarkable levels of genetic diversity. The spatial structure of this diversity was found to be mainly explained by the forest cover of the landscape, suggesting that the landscape has been composed by patches of forests and grasslands for a long time. The main role of forest cover for the connectivity among populations also highlights the importance of riparian forest as dispersal corridors. Finally, differences between organelle and nuclear markers were not enough to establish any strong conclusion about the differences between pollen and seed dispersal. The results presented by Jordi Salmona and collaborators [4] contribute to the understanding of the history and ecology of understudied Madagascar ecosystems. Previous population genetic studies in some forest-dwelling mammals have been interpreted as supporting an old age for the fragmented landscapes in northern Madagascar [e.g. 1,6]. To my knowledge, this is the first study on a tree species. While this work might not completely settle the debate, it emphasizes the importance of studying a diversity of species to understand the biogeographic dynamics of a region. References 1. Quéméré, E., X. Amelot, J. Pierson, B. Crouau-Roy, L. Chikhi (2012) Genetic data suggest a natural prehuman origin of open habitats in northern Madagascar and question the deforestation narrative in this region. Proceedings of the National Academy of Sciences of the United States of | How ancient forest fragmentation and riparian connectivity generate high levels of genetic diversity in a micro-endemic Malagasy tree | Jordi Salmona, Axel Dresen, Anicet E. Ranaivoson, Sophie Manzi, Barbara Le Pors, Cynthia Hong-Wa, Jacqueline Razanatsoa, Nicole V. Andriaholinirina, Solofonirina Rasoloharijaona, Marie-Elodie Vavitsara, Guillaume Besnard | <p>Understanding landscape changes is central to predicting evolutionary trajectories and defining conservation practices. While human-driven deforestation is intense throughout Madagascar, exception in areas like the Loky-Manambato region (North)... | | Evolutionary Ecology, Phylogeography & Biogeography, Population Genetics / Genomics | Miguel de Navascués | 2020-11-27 09:07:21 | ||
04 Sep 2019

The discernible and hidden effects of clonality on the genotypic and genetic states of populations: improving our estimation of clonal ratesHow to estimate clonality from genetic data: use large samples and consider the biology of the speciesRecommended by Myriam Heuertz based on reviews by David Macaya-Sanz, Marcela Van Loo and 1 anonymous reviewer
Population geneticists frequently use the genetic and genotypic information of a population sample of individuals to make inferences on the reproductive system of a species. The detection of clones, i.e. individuals with the same genotype, can give information on whether there is clonal (vegetative) reproduction in the species. If clonality is detected, population geneticists typically use genotypic richness R, the number of distinct genotypes relative to the sample size, to estimate the rate of clonality c, which can be defined as the proportion of reproductive events that are clonal. Estimating the rate of clonality based on genotypic richness is however problematic because, to date, there is no analytical, nor simulation-based, characterization of this relationship. Furthermore, the effect of sampling on this relationship has never been critically examined. References [1] Stoeckel, S., Porro, B., and Arnaud-Haond, S. (2019). The discernible and hidden effects of clonality on the genotypic and genetic states of populations: improving our estimation of clonal rates. ArXiv:1902.09365 [q-Bio] v4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. Retrieved from http://arxiv.org/abs/1902.09365v4 | The discernible and hidden effects of clonality on the genotypic and genetic states of populations: improving our estimation of clonal rates | Solenn Stoeckel, Barbara Porro, Sophie Arnaud-Haond | <p>Partial clonality is widespread across the tree of life, but most population genetics models are conceived for exclusively clonal or sexual organisms. This gap hampers our understanding of the influence of clonality on evolutionary trajectories... | | Population Genetics / Genomics, Reproduction and Sex | Myriam Heuertz | 2019-02-28 10:10:56 | ||
05 Apr 2024
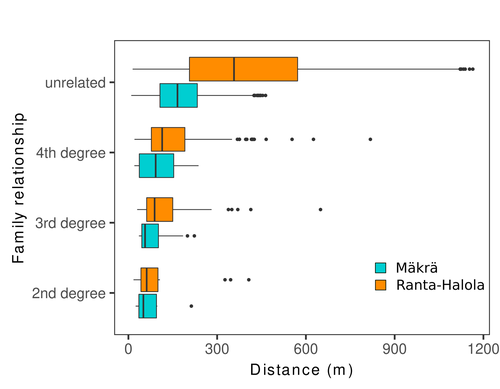
Does the seed fall far from the tree? Weak fine scale genetic structure in a continuous Scots pine populationWeak spatial genetic structure in a large continuous Scots pine population – implications for conservation and breedingRecommended by Myriam Heuertz based on reviews by Joachim Mergeay, Jean-Baptiste Ledoux and Roberta Loh
Spatial genetic structure, i.e. the non-random spatial distribution of genotypes, arises in populations because of different processes including spatially limited dispersal and selection. Knowledge on the spatial genetic structure of plant populations is important to assess biological parameters such as gene dispersal distances and the potential for local adaptations, as well as for applications in conservation management and breeding. In their work, Niskanen and colleagues demonstrate a multifaceted approach to characterise the spatial genetic structure in two replicate sites of a continuously distributed Scots pine population in South-Eastern Finland. They mapped and assessed the ages of 469 naturally regenerated adults and genotyped them using a SNP array which resulted in 157 325 filtered polymorphic SNPs. Their dataset is remarkably powerful because of the large numbers of both individuals and SNPs genotyped. This made it possible to characterise precisely the decay of genetic relatedness between individuals with spatial distance despite the extensive dispersal capacity of Scots pine through pollen, and ensuing expectations of an almost panmictic population. The authors’ data analysis was particularly thorough. They demonstrated that two metrics of pairwise relatedness, the genomic relationship matrix (GRM, Yang et al. 2011) and the kinship coefficient (Loiselle et al. 1995) were strongly correlated and produced very similar inference of family relationships: >99% of pairs of individuals were unrelated, and the remainder exhibited 2nd (e.g., half-siblings) to 4th degree relatedness. Pairwise relatedness decayed with spatial distance which resulted in extremely weak but statistically significant spatial genetic structure in both sites, quantified as Sp=0.0005 and Sp=0.0008. These estimates are at least an order of magnitude lower than estimates in the literature obtained in more fragmented populations of the same species or in other conifers. Estimates of the neighbourhood size, the effective number of potentially mating individuals belonging to a within-population neighbourhood (Wright 1946), were relatively large with Nb=1680-3210 despite relatively short gene dispersal distances, σg = 36.5–71.3m, which illustrates the high effective density of the population. The authors showed the implications of their findings for selection. The capacity for local adaptation depends on dispersal distances and the strength of the selection coefficient. In the study population, the authors inferred that local adaptation can only occur if environmental heterogeneity occurs over a distance larger than approximately one kilometre (or larger, if considering long-distance dispersal). Interestingly, in Scots pine, no local adaptation has been described on similar geographic scales, in contrast to some other European or Mediterranean conifers (Scotti et al. 2023). The authors’ results are relevant for the management of conservation and breeding. They showed that related individuals occurred within sites only and that they shared a higher number of rare alleles than unrelated ones. Since rare alleles are enriched in new and recessive deleterious variants, selecting related individuals could have negative consequences in breeding programmes. The authors also showed, in their response to reviewers, that their powerful dataset was not suitable to obtain a robust estimate of effective population size, Ne, based on the linkage disequilibrium method (Do et al. 2014). This illustrated that the estimation of Ne used for genetic indicators supported in international conservation policy (Hoban et al. 2020, CBD 2022) remains challenging in large and continuous populations (see also Santo-del-Blanco et al. 2023, Gargiulo et al. 2024). ReferencesCBD (2022) Kunming-Montreal Global Biodiversity Framework. https://www.cbd.int/doc/decisions/cop-15/cop-15-dec-04-en.pdf Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014). NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne ) from genetic data. Molecular Ecology Resources 14: 209–214. https://doi.org/10.1111/1755-0998.12157 Gargiulo R, Decroocq V, González-Martínez SC, Paz-Vinas I, Aury JM, Kupin IL, Plomion C, Schmitt S, Scotti I, Heuertz M (2024) Estimation of contemporary effective population size in plant populations: limitations of genomic datasets. Evolutionary Applications, in press, https://doi.org/10.1101/2023.07.18.549323 Hoban S, Bruford M, D’Urban Jackson J, Lopes-Fernandes M, Heuertz M, Hohenlohe PA, Paz-Vinas I, et al. (2020) Genetic diversity targets and indicators in the CBD post-2020 Global Biodiversity Framework must be improved. Biological Conservation 248: 108654. https://doi.org/10.1016/j.biocon.2020.108654 Loiselle BA, Sork VL, Nason J & Graham C (1995) Spatial genetic structure of a tropical understorey shrub, Psychotria officinalis (Rubiaceae). American Journal of Botany 82: 1420–1425. https://doi.org/10.1002/j.1537-2197.1995.tb12679.x Santos-del-Blanco L, Olsson S, Budde KB, Grivet D, González-Martínez SC, Alía R, Robledo-Arnuncio JJ (2022). On the feasibility of estimating contemporary effective population size (Ne) for genetic conservation and monitoring of forest trees. Biological Conservation 273: 109704. https://doi.org/10.1016/j.biocon.2022.109704 Scotti I, Lalagüe H, Oddou-Muratorio S, Scotti-Saintagne C, Ruiz Daniels R, Grivet D, et al. (2023) Common microgeographical selection patterns revealed in four European conifers. Molecular Ecology 32: 393-411. https://doi.org/10.1111/mec.16750 Wright S (1946) Isolation by distance under diverse systems of mating. Genetics 31: 39–59. https://doi.org/10.1093/genetics/31.1.39 Yang J, Lee SH, Goddard ME & Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. The American Journal of Human Genetics 88: 76–82. https://www.cell.com/ajhg/pdf/S0002-9297(10)00598-7.pdf | Does the seed fall far from the tree? Weak fine scale genetic structure in a continuous Scots pine population | Alina K. Niskanen, Sonja T. Kujala, Katri Kärkkäinen, Outi Savolainen, Tanja Pyhäjärvi | <p>Knowledge of fine-scale spatial genetic structure, i.e., the distribution of genetic diversity at short distances, is important in evolutionary research and in practical applications such as conservation and breeding programs. In trees, related... | | Adaptation, Evolutionary Applications, Population Genetics / Genomics | Myriam Heuertz | Joachim Mergeay | 2023-06-27 21:57:28 |




