
Latest recommendations

| Id | Title | Authors▼ | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
01 Sep 2021

Connectivity and selfing drives population genetic structure in a patchy landscape: a comparative approach of four co-occurring freshwater snail speciesDeterminants of population genetic structure in co-occurring freshwater snailsRecommended by Trine Bilde and Matteo Fumagalli based on reviews by 3 anonymous reviewers and Matteo Fumagalli based on reviews by 3 anonymous reviewers
Genetic diversity is a key aspect of biodiversity and has important implications for evolutionary potential and thereby the persistence of species. Improving our understanding of the factors that drive genetic structure within and between populations is, therefore, a long-standing goal in evolutionary biology. However, this is a major challenge, because of the complex interplay between genetic drift, migration, and extinction/colonization dynamics on the one hand, and the biology and ecology of species on the other hand (Romiguier et al. 2014, Ellegren and Galtier 2016, Charlesworth 2003). Jarne et al. (2021) studied whether environmental and demographic factors affect the population genetic structure of four species of hermaphroditic freshwater snails in a similar way, using comparative analyses of neutral genetic microsatellite markers. Specifically, they investigated microsatellite variability of Hygrophila in almost 280 sites in Guadeloupe, Lesser Antilles, as part of a long-term survey experiment (Lamy et al. 2013). They then modelled the influence of the mating system, local environmental characteristics and demographic factors on population genetic diversity. Consistent with theoretical predictions (Charlesworth 2003), they detected higher genetic variation in two outcrossing species than in two selfing species, emphasizing the importance of the mating system in maintaining genetic diversity. The study further identified an important role of site connectivity, through its influences on effective population size and extinction/colonisation events. Finally, the study detects an influence of interspecific interactions caused by an ongoing invasion by one of the studied species on genetic structure, highlighting the indirect effect of changes in community composition and demography on population genetics. Jarne et al. (2021) could address the extent to which genetic structure is determined by demographic and environmental factors in multiple species given the remarkable sampling available. Additionally, the study system is extremely suitable to address this hypothesis as species’ habitats are defined and delineated. Whilst the authors did attempt to test for across-species correlations, further investigations on this matter are required. Moreover, the effect of interactions between factors should be appropriately considered in any modelling between genetic structure and local environmental or demographic features. The findings in this study contribute to improving our understanding of factors influencing population genetic diversity, and highlights the complexity of interacting factors, therefore also emphasizing the challenges of drawing general implications, additionally hampered by the relatively limited number of species studied. Jarne et al. (2021) provide an excellent showcase of an empirical framework to test determinants of genetic structure in natural populations. As such, this study can be an example for further attempts of comparative analysis of genetic diversity. References Charlesworth, D. (2003) Effects of inbreeding on the genetic diversity of populations. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences, 358, 1051-1070. doi: https://doi.org/10.1098/rstb.2003.1296 Ellegren, H. and Galtier, N. (2016) Determinants of genetic diversity. Nature Reviews Genetics, 17, 422-433. doi: https://doi.org/10.1038/nrg.2016.58 Jarne, P., Lozano del Campo, A., Lamy, T., Chapuis, E., Dubart, M., Segard, A., Canard, E., Pointier, J.-P. and David, P. (2021) Connectivity and selfing drives population genetic structure in a patchy landscape: a comparative approach of four co-occurring freshwater snail species. HAL, hal-03295242, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://hal.archives-ouvertes.fr/hal-03295242 Lamy, T., Gimenez, O., Pointier, J. P., Jarne, P. and David, P. (2013). Metapopulation dynamics of species with cryptic life stages. The American Naturalist, 181, 479-491. doi: https://doi.org/10.1086/669676 Romiguier, J., Gayral, P., Ballenghien, M. et al. (2014) Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature, 515, 261-263. doi: https://doi.org/10.1038/nature13685 | Connectivity and selfing drives population genetic structure in a patchy landscape: a comparative approach of four co-occurring freshwater snail species | Jarne P., Lozano del Campo A., Lamy T., Chapuis E., Dubart M., Segard A., Canard E., Pointier J.-P., David P. | <p style="text-align: justify;">The distribution of neutral genetic variation in subdivided populations is driven by the interplay between genetic drift, migration, local extinction and colonization. The influence of environmental and demographic ... | | Adaptation, Evolutionary Dynamics, Population Genetics / Genomics, Reproduction and Sex, Species interactions | Trine Bilde | 2021-02-11 19:57:51 | ||
05 Jun 2018
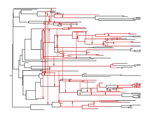
The dynamics of preferential host switching: host phylogeny as a key predictor of parasite prevalence and distributionShift or stick? Untangling the signatures of biased host switching, and host-parasite co-speciationRecommended by Lucy Weinert based on reviews by Damien de Vienne and Nathan MeddMany emerging diseases arise by parasites switching to new host species, while other parasites seem to remain with same host lineage for very long periods of time, even over timescales where an ancestral host species splits into two or more new species. The ability to understand these dynamics would form an important part of our understanding of infectious disease. Experiments are clearly important for understanding these processes, but so are comparative studies, investigating the variation that we find in nature. Such comparative data do show strong signs of non-randomness, and this suggests that the epidemiological and ecological processes might be predictable, at least in part. For example, when we map patterns of parasite presence/absence onto host phylogenies, we often find that certain host clades harbour many more parasites than expected, or that closely-related hosts harbour closely-related parasites. Nevertheless, it remains difficult to interpret these patterns to make inferences about ecological and epidemiological processes. This is partly because non-random associations can arise in multiple ways. For example, parasites might be inherited from the common ancestor of related hosts, or might switch to new hosts, but preferentially establish on novel hosts that are closely related to their existing host. Infection might also influence the shape of host phylogeny, either by increasing the rate of host extinction or, conversely, increasing the rate of speciation (as with manipulative symbionts that might induce reproductive isolation). These various processes have, by and large, been studied in isolation, but the model introduced by Engelstädter and Fortuna [1], makes an important first step towards studying them together. Without such combined analyses, we will not be able to tell if the processes have their own unique signatures, or whether the same sort of non-randomness can arise in multiple ways. A major finding of the work is that the size of a host clade can be an important determinant of its overall infection level. This had been shown in previous work, assuming that the host phylogeny was fixed, but the current paper shows that it extends also to situations where host extinction and speciation takes place at a comparable rate to host shifting. This finding, then, calls into question the natural assumption that a clade of host species that is highly parasite ridden, must have some genetic or ecological characteristic that makes them particularly prone to infection, arguing that the clade size, rather than any characteristic of the clade members, might be the important factor. It will be interesting to see whether this prediction about clade size is borne out with comparative studies. Another feature of the study is that the framework is naturally extendable, to include further processes, such as the influence of parasite presence on extinction or speciation rates. No doubt extensions of this kind will form the basis of important future work. References [1] Engelstädter J and Fortuna NZ. 2018. The dynamics of preferential host switching: host phylogeny as a key predictor of parasite prevalence and distribution. bioRxiv 209254 ver. 5 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/209254 | The dynamics of preferential host switching: host phylogeny as a key predictor of parasite prevalence and distribution | Jan Engelstaedter & Nicole Fortuna | <p>New parasites commonly arise through host-shifts, where parasites from one host species jump to and become established in a new host species. There is much evidence that the probability of host-shifts decreases with increasing phylogenetic dist... | | Bioinformatics & Computational Biology, Evolutionary Epidemiology, Evolutionary Theory, Macroevolution, Phylogenetics / Phylogenomics, Species interactions | Lucy Weinert | 2017-10-30 02:06:06 | ||
29 Nov 2023
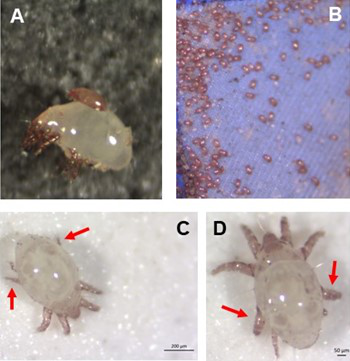
Individual differences in developmental trajectory leave a male polyphenic signature in bulb mite populationsWhat determines whether to scramble or fight in male bulb mitesRecommended by Ulrich Karl Steiner based on reviews by 2 anonymous reviewers
A classic textbook example in evolutionary ecology for phenotypic plasticity—the expression of different phenotypes by a genotype under different environmental conditions—is on Daphnia (Dodson 1989). If various species of this small crustacean are exposed to predation risk or cues thereof, their offspring show induced defense phenotypes including helmets, neck teeth, or head- and tail-spines. These induced morphological changes lower the risk of being eaten by a predator. As in Daphnia, induction can span over generations, while other induced phenotypic plastic changes are almost instantaneous, including many responses in physiology and behaviour (Gabriel et al. 2005). Larvae male bulb mites also show plasticity in morphologies throughout development. They can develop into a costly adult fighter morphology or a less costly, but vulnerable, scrambler type. The question Deere and Smallegange (Deere & Smallegange 2023) address is whether male bulb mite larvae can anticipate which type will likely be adaptive once they become adult, or alternatively, whether the resource availability or population density they experience during their larvae phase determines frequencies of adult scrambler and fighter types. They explore this question through experimental evolution, by removing different fractions of developing intermediate larva types. They thereby manipulate the stage structure of populations and alter selective forces on these stages. The potential shift of fixed genetics, imposed by the experimental selection regimes, is evaluated by fitness assays in green garden experiments. The exciting extension to classical experiments on phenotypic plasticity is that the authors aim at exploring eco-evolutionary feedbacks experimentally in a system that is a little more complex than basic host-parasite or predator-prey systems. The latter involve, for instance, rotifer-algae dynamics (Yoshida et al. 2003; Becks et al. 2012) or similar simple lab systems for which eco-evolutionary feedbacks have been demonstrated. The challenge for the exploration of more complex systems is revealed in the study by Deere and Smallegange. Their findings suggest that the frequencies of adult male morphotype is triggered by the environmental condition (nutrient availability) during the larval phase, i.e. a simple environmental induced plastic response. No fixed genetic shift in determining adult morphotype frequencies occurs. The trigger at the larva phase remains also not perfectly determined in their experiments, as population density and resource (food) availability are partly confounded. Additional complexity and selective aspects come into play in this system, as the targeted developmental stages that develop into fighter male morphs are also dispersal morphs. If selection on dispersal to avoid residing in food-limited environments is strong, triggering genetic shifts in fighter morphs by local population structure might be hard to experimentally achieve. Small sample sizes limit conclusions on complex interactions among duration of the experiment, population size, developmental stage types, and adult fighter frequencies. The presented study (Deere & Smallegange 2023) helps to bridge theoretical predictions and empirical evidence for eco-evolutionary feedbacks that goes beyond simple ecological-driven changes in population dynamics (Govaert et al. 2019).
References
Becks, L., Ellner, S.P., Jones, L.E. & Hairston, N.G. (2012). The functional genomics of an eco-evolutionary feedback loop: linking gene expression, trait evolution, and community dynamics. Ecol. Lett., 15, 492-501. | Individual differences in developmental trajectory leave a male polyphenic signature in bulb mite populations | Jacques A. Deere & Isabel M. Smallegange | <p style="text-align: justify;">Developmental plasticity alters phenotypes and can in that way change the response to selection. When alternative phenotypes show different life history trajectories, developmental plasticity can also affect, and be... | | Evolutionary Ecology, Life History, Phenotypic Plasticity, Sexual Selection | Ulrich Karl Steiner | 2023-02-07 12:14:33 | ||
13 Dec 2016

POSTPRINT
Repeated replacements of an intrabacterial symbiont in the tripartite nested mealybug symbiosisObligate dependence does not preclude changing partners in a Russian dolls symbiotic systemRecommended by Emmanuelle Jousselin and Fabrice VavreSymbiotic associations with bacterial partners have facilitated important evolutionary transitions in the life histories of eukaryotes. For instance, many insects have established long-term interactions with intracellular bacteria that provide them with essential nutrients lacking in their diet. However, despite the high level of interdependency among organisms involved in endosymbiotic systems, examples of symbiont replacements along the evolutionary history of insect hosts are numerous.
In their paper, Husnik and McCutcheon [1] test the stability of symbiotic systems in a particularly imbricated Russian-doll type interaction, where one bacterium lives insides another bacterium, which itself lives inside insect cells. For their study, they chose representative species of mealybugs (Pseudococcidae), a species rich group of sap-feeding insects that hosts diverse and complex symbiotic systems. In species of the subfamily Pseudococcinae, data published so far suggest that the primary symbiont, a ß-proteobacterium named Tremblaya princeps, is supplemented by a second bacterial symbiont (a ϒ-proteobaterium) that lives within its cytoplasm; both participate to the metabolic pathways that provide essential amino acids and vitamins to their hosts. Here, Husnik and McCutcheon generate host and endosymbiont genome data for five phylogenetically divergent species of Pseudococcinae in order to better understand: 1) the evolutionary history of the symbiotic associations; 2) the metabolic roles of each partner, 3) the timing and origin of Horizontal Gene Transfers (HGT) between the hosts and their symbionts. Reference [1] Husnik F., McCutcheon JP. 2016. Repeated replacements of an intrabacterial symbiont in the tripartite nested mealybug symbiosis. PNAS 113: E5416-E5424. doi: 10.1073/pnas.1603910113 | Repeated replacements of an intrabacterial symbiont in the tripartite nested mealybug symbiosis | Husnik F, McCutcheon JP | Stable endosymbiosis of a bacterium into a host cell promotes cellular and genomic complexity. The mealybug *Planococcus citri* has two bacterial endosymbionts with an unusual nested arrangement: the γ-proteobacterium *Moranella endobia* lives in ... | | Phylogenetics / Phylogenomics, Species interactions | Emmanuelle Jousselin | 2016-12-13 14:27:09 | ||
25 Jan 2023
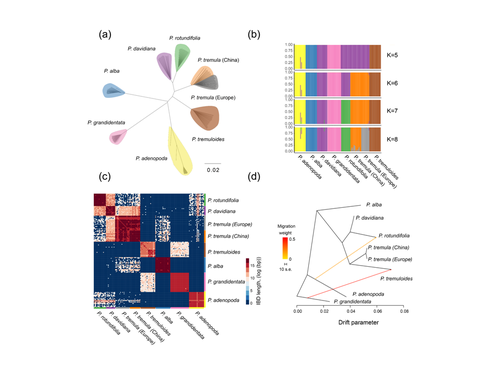
Drivers of genomic landscapes of differentiation across Populus divergence gradientShedding light on genomic divergence along the speciation continuumRecommended by Violaine Llaurens based on reviews by Camille Roux, Steven van Belleghem and 1 anonymous reviewer
The article “Drivers of genomic landscapes of differentiation across Populus divergence gradient” by Shang et al. describes an amazing dataset where genomic variations among 21 pairs of diverging poplar species are compared. Such comparisons are still quite rare and are needed to shed light on the processes shaping genomic divergence along the speciation gradient. Relying on two hundred whole-genome resequenced samples from 8 species that diverged from 1.3 to 4.8 million years ago, the authors aim at identifying the key factors involved in the genomic differentiation between species. They carried out a wide range of robust statistical tests aiming at characterizing the genomic differentiation along the genome of these species pairs. They highlight in particular the role of linked selection and gene flow in shaping the divergence along the genomes of species pairs. They also confirm the significance of introgression among species with a net divergence larger than the upper boundaries of the grey zone of speciation previously documented in animals (da from 0.005 to 0.02, Roux et al. 2016). Because these findings pave the way to research about the genomic mechanisms associated with speciation in species with allopatric and parapatric distributions, I warmingly recommend this article. References Roux C, Fraïsse C, Romiguier J, Anciaux Y, Galtier N, Bierne N (2016) Shedding Light on the Grey Zone of Speciation along a Continuum of Genomic Divergence. PLOS Biology, 14, e2000234. https://doi.org/10.1371/journal.pbio.2000234 Shang H, Rendón-Anaya M, Paun O, Field DL, Hess J, Vogl C, Liu J, Ingvarsson PK, Lexer C, Leroy T (2023) Drivers of genomic landscapes of differentiation across Populus divergence gradient. bioRxiv, 2021.08.26.457771, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.08.26.457771 | Drivers of genomic landscapes of differentiation across Populus divergence gradient | Huiying Shang, Martha Rendón-Anaya, Ovidiu Paun, View David L Field, Jaqueline Hess, Claus Vogl, Jianquan Liu, Pär K. Ingvarsson, Christian Lexer, Thibault Leroy | <p style="text-align: justify;">Speciation, the continuous process by which new species form, is often investigated by looking at the variation of nucleotide diversity and differentiation across the genome (hereafter genomic landscapes). A key cha... | | Population Genetics / Genomics, Speciation | Violaine Llaurens | 2021-09-06 14:12:27 | ||
02 May 2023
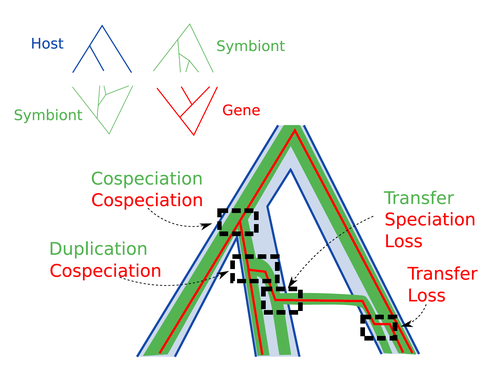
Host-symbiont-gene phylogenetic reconciliationReconciling molecular evolution and evolutionary ecology studies: a phylogenetic reconciliation method for gene-symbiont-host systemsRecommended by Emmanuelle Jousselin based on reviews by Vincent Berry and Catherine MatiasInteractions between species are a driving force in evolution. Many organisms host symbiotic partners that live all or part of their life in or on their host. Whether they are mutualistic or parasitic, these symbiotic associations impose strong selective pressures on both partners and affect their evolutionary trajectories. In fine, they can have a significant impact on the diversification patterns of both host and symbiont lineages, with symbiotic lineages sometimes speciating simultaneously with their hosts and/or switching from one host species to another. Long-term associations between species can also result in gene transfers between the involved organisms. Those lateral gene transfers are a source of ecological innovation but can obscure the phylogenetic signals and render the process of phylogenetic reconstructions complex (Lerat et al. 2003). Methods known as reconciliations explore similarities and differences between phylogenetic trees. They have been widely used to both compare the diversification patterns of hosts and symbionts and identify lateral gene transfers between species. Though the reconciliation approaches used in host/ symbiont and species/ gene phylogenetic studies are identical, they are always applied separately to solve either molecular evolution questions or investigate the evolution of ecological interactions. However, the two questions are often intimately linked and the current interest in multi-level systems (e.g. the holobiont concept) calls for a unique model that will take into account three-level nested organization (gene/symbiont/ host) where both symbiont and genes can transfer among hosts. Here Menet and collaborators (2023) provide such a model to produce three-level reconciliations. In order to do so, they extend the two-level reconciliation model implemented in “ALE” software (Szöllősi et al. 2013), one of the most used and proven reconciliation methods. Briefly, given a symbiont gene tree, a symbiont tree and a host tree, as in previous reconciliation models, the symbiont tree is mapped onto the host tree by mixing three types of events: Duplication, Transfer or Loss (DTL), with a possibility of the symbiont evolving temporarily outside the host phylogeny (in a “ghost” host lineage). The gene tree evolves similarly inside the symbiont tree, but horizontal transfers are constrained to symbionts co-occurring within the same host. Joint reconciliation scenarios are reconstructed and DTL event rates and likelihoods are estimated according to the model. As a nice addition, the authors propose a method to infer the symbiont phylogeny through amalgamation from gene trees and a host tree. The authors then explore the diverse possibilities offered by this method by testing it on both simulated datasets and biological datasets in order to check whether considering three nested levels is worthwhile. They convincingly show that three-level reconciliation has a better capacity to retrieve the symbiont donors and receivers of horizontal gene transfers, probably because transfers are constrained by additional elements relevant to the biological systems. Using, aphids, their obligate endosymbionts, and the symbiont genes involved in their nutritional functions, they identify horizontal gene transfers between aphid symbionts that are missed by two-level reconciliations but detected by expertise (Manzano-Marín et al. 2020). The other dataset presented here is on the human pathogen Helicobacter pylori, which history is supposed to reflect human migration. They use more than 1000 H. pylori gene families, and four populations, and use likelihood computations to compare different hypotheses on the diversification of the host. In summary, this study is a proof-of-concept of a 3-level reconciliation, where the authors manage to convey the applicability of their framework to many biological systems. Reported complexities, confirmed by reported running times, show that the method is computationally efficient. Without a doubt, the tool presented here will be very useful to evolutionary biologists who want to investigate multi-scale cophylogenies and it will move forward the study of associations between host and symbionts when symbiont genomic data are available. REFERENCES Lerat, E., Daubin, V., & Moran, N. A. (2003). From gene trees to organismal phylogeny in prokaryotes: the case of the γ-Proteobacteria. PLoS biology, 1(1), e19. Menet H, Trung AN, Daubin V, Tannier E (2023) Host-symbiont-gene phylogenetic reconciliation. bioRxiv, 2022.07.01.498457, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.07.01.498457 Szöllősi, G. J., Rosikiewicz, W., Boussau, B., Tannier, E., & Daubin, V. (2013). Efficient exploration of the space of reconciled gene trees. Systematic biology, 62(6), 901-912. | Host-symbiont-gene phylogenetic reconciliation | Hugo Menet, Alexia Nguyen Trung, Vincent Daubin, Eric Tannier | <p style="text-align: justify;"><strong>Motivation:</strong> Biological systems are made of entities organized at different scales e.g. macro-organisms, symbionts, genes...) which evolve in interaction.<br>These interactions range from indepe... | | Bioinformatics & Computational Biology, Phylogenetics / Phylogenomics | Emmanuelle Jousselin | 2022-08-21 18:34:27 | ||
20 Nov 2019
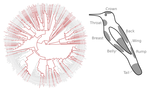
Distribution of iridescent colours in hummingbird communities results from the interplay between selection for camouflage and communicationFeathers iridescence sheds light on the assembly rules of humingbirds communitiesRecommended by Sébastien Lavergne based on reviews by 2 anonymous reviewersEcology needs rules stipulating how species distributions and ecological communities should be assembled along environmental gradients, but few rules have yet emerged in the ecological literature. The search of ecogeographical rules governing the spatial variation of birds colours has recently known an upsurge of interest in the litterature [1]. Most studies have, however, looked at pigmentary colours and not structural colours (e.g. iridescence), although it is know that color perception by animals (both birds and their predators) can be strongly influenced by light diffraction causing iridescence patterns on feathers. References [1] Delhey, K. (2019). A review of Gloger’s rule, an ecogeographical rule of colour: definitions, interpretations and evidence. Biological Reviews, 94(4), 1294–1316. doi: 10.1111/brv.12503 | Distribution of iridescent colours in hummingbird communities results from the interplay between selection for camouflage and communication | Hugo Gruson, Marianne Elias, Juan L. Parra, Christine Andraud, Serge Berthier, Claire Doutrelant, Doris Gomez | <p>Identification errors between closely related, co-occurring, species may lead to misdirected social interactions such as costly interbreeding or misdirected aggression. This selects for divergence in traits involved in species identification am... | | Evolutionary Ecology, Macroevolution, Phylogeography & Biogeography, Sexual Selection, Species interactions | Sébastien Lavergne | 2019-03-29 17:23:20 | ||
20 Dec 2016

POSTPRINT
Experimental Evolution of Gene Expression and Plasticity in Alternative Selective RegimesGenetic adaptation counters phenotypic plasticity in experimental evolutionRecommended by Luis-Miguel Chevin and Stephanie BedhommeHow do phenotypic plasticity and adaptive evolution interact in a novel or changing environment? Does evolution by natural selection generally reinforce initially plastic phenotypic responses, or does it instead oppose them? And to what extent does evolution of a trait involve evolution of its plasticity? These questions have lied at the heart of research on phenotypic evolution in heterogeneous environments ever since it was realized that the environment is likely to affect the expression of many (perhaps most) characters of an individual. Importantly, this broad definition of phenotypic plasticity as change in the average phenotype of a given genotype in response to its environment of development (or expression) does not involve any statement about the adaptiveness of the plastic changes. Theory on the evolution of plasticity has devoted much effort to understanding how reaction norm should evolve under different regimes of environmental change in space and time, and depending on genetic constraints on reaction norm shapes. However on an empirically ground, the questions above have mostly been addressed for individual traits, often chosen a priori for their likeliness to exhibit adaptive plasticity, and we still lack more systematic answers. These can be provided by so-called ‘phenomic’ approaches, where a large number of traits are tracked without prior information on their biological or ecological function. A problem is that the number of phenotypic characters that can be measured in an organism is virtually infinite (and to some extent arbitrary), and that scaling issues makes it difficult to compare different sets of traits. Gene-expression levels offer a partial solution to this dilemma, as they can be considered as a very large number of traits (one per typed gene) that can be measured easily and uniformly (fold change in the number of reads in RNAseq). As for any traits, expression levels of different genes may be genetically correlated, to an extent that depends on their regulation mechanism: cis-regulatory sequences that only affect expression of neighboring genes are likely to cause independent gene expression, while more systematic modifiers of expression (e.g. trans-regulators such as transcription factors) may cause correlated genetic responses of the expression of many genes. Huang and Agrawal [1] have studied plasticity and evolution of gene expression level in young larvae of populations of Drosophila melanogaster that have evolved for about 130 generations under either a constant environment (salt or cadmium), or an environment that is heterogeneous in time or space (combining salt and cadmium). They report a wealth of results, of which we summarize the most striking here. First, among genes that (i) were initially highly plastic and (ii) evolved significant divergence in expression levels between constant environment treatments, the evolved divergence is predominantly in the opposite direction to the initial plastic response. This suggests that either plasticity was initially maladaptive, or the selective pressure changed during the evolutionary process (see below). This somewhat unexpected result strikingly mirrors that from a study published last year in Nature [2], where the same pattern was found for responses of guppies to the presence of predators. However, Huang and Agrawal [1] went beyond this study by deciphering the underlying mechanisms in several interesting ways. First, they showed that change in gene expression often occurred at genes close to SNPs with differentiated frequencies across treatments (but not at genes with differentiated SNPs in their coding sequences), suggesting that cis-regulatory sequences are involved. This is also suggested by the fact that changes in gene expression are mostly caused by the increased expression of only one allele at polymorphic loci, and is a first step towards investigating the genetic underpinnings of (co)variation in gene expression levels. Another interesting set of findings concerns evolution of plasticity in treatments with variable environments. To compare the gene-expression plasticity that evolved in these treatments to an expectation, the authors considered that the expression levels in populations maintained for a long time under constant salt or cadmium had reached an optimum. The differences between these expression levels were thus assumed to predict the level of plasticity that should evolve in a heterogeneous environment (with both cadmium and salt) under perfect environmental predictability. The authors showed that plasticity did evolve more in the expected direction in heterogeneous than in constant environments, resulting in better adapted final expression levels across environments. Taken collectively, these results provide an unprecedented set of patterns that are greatly informative on how plasticity and evolution interact in constant versus changing environments. But of course, interpretations in terms of adaptive versus maladaptive plasticity are more challenging, as the authors themselves admit. Even though environmentally determined gene expression is the basic mechanism underlying the phenotypic plasticity of most traits, it is extremely difficult to relate to more integrated phenotypes for which we can understand the selection pressures, especially in multicellular organisms. The authors have recently investigated evolutionary change of quantitative traits in these selected lines, so it might be possible to establish links between reaction norms for macroscopic traits to those for gene expression levels. Such an approach would also involve tracking gene expression throughout life, rather than only in young larvae as done here, thus putting phenotypic complexity back in the picture also for expression levels. Another difficulty is that a plastic response that was originally adaptive may be replaced by an opposite evolutionary response in the long run, without having to invoke initially maladaptive plasticity. For instance, the authors mention the possibility that a generic stress response is initially triggered by cadmium, but is eventually unnecessary and costly after evolution of genetic mechanisms for cadmium detoxification (a case of so-called genetic accommodation). In any case, this study by Huang and Agrawal [1], together with the one by Ghalambor et al. last year [2], reports novel and unexpected results, which are likely to stimulate researchers interested in plasticity and evolution in heterogeneous environments for the years to come. References [1] Huang Y, Agrawal AF. 2016. Experimental Evolution of Gene Expression and Plasticity in Alternative Selective Regimes. PLoS Genetics 12:e1006336. doi: 10.1371/journal.pgen.1006336 [2] Ghalambor CK, Hoke KL, Ruell EW, Fischer EK, Reznick DN, Hughes KA. 2015. Non-adaptive plasticity potentiates rapid adaptive evolution of gene expression in nature. Nature 525: 372-375. doi: 10.1038/nature15256 | Experimental Evolution of Gene Expression and Plasticity in Alternative Selective Regimes | Huang Y, Agrawal AF | Little is known of how gene expression and its plasticity evolves as populations adapt to different environmental regimes. Expression is expected to evolve adaptively in all populations but only those populations experiencing environmental heterog... | | Adaptation, Experimental Evolution, Expression Studies, Phenotypic Plasticity | Luis-Miguel Chevin | 2016-12-20 09:04:15 | ||
17 Dec 2016
POSTPRINT
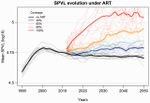
Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling studyPredicting HIV virulence evolution in response to widespread treatmentRecommended by Samuel Alizon and Roger Kouyos
It is a classical result in the virulence evolution literature that treatments decreasing parasite replication within the host should select for higher replication rates, thus driving increased levels of virulence if the two are correlated. There is some evidence for this in vitro but very little in the field. HIV infections in humans offer a unique opportunity to go beyond the simple predictions that treatments should favour more virulent strains because many details of this host-parasite system are known, especially the link between set-point virus load, transmission rate and virulence. To tackle this question, Herbeck et al. [1] used a detailed individual-based model. This is original because it allows them to integrate existing knowledge from the epidemiology and evolution of HIV (e.g. recent estimates of the ‘heritability’ of set-point virus load from one infection to the next). This detailed model allows them to formulate predictions regarding the effect of different treatment policies; especially regarding the current policy switch away from treatment initiation based on CD4 counts towards universal treatment. The results show that, perhaps as expected from the theory, treatments based on the level of remaining host target cells (CD4 T cells) do not affect virulence evolution because they do not strongly affect the virulence level that maximizes HIV’s transmission potential. However, early treatments can lead to moderate increase in virulence within several years if coverage is high enough. These results seem quite robust to variation of all the parameters in realistic ranges. The great step forward in this model is the ability to obtain quantitative prediction regarding how a virus may evolve in response to public health policies. Here the main conclusion is that given our current knowledge in HIV biology, the risk of virulence evolution is perhaps more limited than expected from a direct application of virulence evolution model. Interestingly, the authors also conclude that recently observed increased in HIV virulence [2-3] cannot be explained by the impact of antiretroviral therapy alone; which raises the question about the main mechanism behind this increase. Finally, the authors make the interesting suggestion that “changing virulence is amenable to being monitored alongside transmitted drug resistance in sentinel surveillance”. References [1] Herbeck JT, Mittler JE, Gottlieb GS, Goodreau SM, Murphy JT, Cori A, Pickles M, Fraser C. 2016. Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling study. Virus Evolution 2:vew028. doi: 10.1093/ve/vew028 [2] Herbeck JT, Müller V, Maust BS, Ledergerber B, Torti C, et al. 2012. Is the virulence of HIV changing? A meta-analysis of trends in prognostic markers of HIV disease progression and transmission. AIDS 26:193-205. doi: 10.1097/QAD.0b013e32834db418 [3] Pantazis N, Porter K, Costagliola D, De Luca A, Ghosn J, et al. 2014. Temporal trends in prognostic markers of HIV-1 virulence and transmissibility: an observational cohort study. Lancet HIV 1:e119-26. doi: 10.1016/s2352-3018(14)00002-2 | Evolution of HIV virulence in response to widespread scale up of antiretroviral therapy: a modeling study | Herbeck JT, Mittler JE, Gottlieb GS, Goodreau SM, Murphy JT, Cori A, Pickles M, Fraser C | There are global increases in the use of HIV antiretroviral therapy (ART), guided by clinical benefits of early ART initiation and the efficacy of treatment as prevention of transmission. Separately, it has been shown theoretically and empirically... | | Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Epidemiology | Samuel Alizon | 2016-12-16 20:54:08 | ||
15 Sep 2022

Bimodal breeding phenology in the Parsley Frog Pelodytes punctatus as a bet-hedging strategy in an unpredictable environment despite strong priority effectsSpreading the risk of reproductive failure when the environment is unpredictable and ephemeralRecommended by Gabriele Sorci based on reviews by Thomas Haaland and Zoltan RadaiMany species breed in environments that are unpredictable, for instance in terms of the availability of resources needed to raise the offspring. Organisms might respond to such spatial and temporal unpredictability by adopting plastic responses to adjust their reproductive investment according to perceived cues of environmental quality. Some species such as the amphibians might also face the problem of ephemeral habitats, when the ponds where they breed have a chance of drying up before metamorphosis has occurred. In this case, maximizing long-term fitness might involve a strategy of spreading the risk, even though the reproductive success of a single reproductive bout might be lower. Understanding how animals (and plants) get adapted to stochastic environments is particularly crucial in the current context of rapid environmental changes. In this article, Jourdan-Pineau et al. report the results of field surveys of the Parsley Frog (Pelodytes punctatus) in Southern France. This frog has peculiar breeding phenology with females breeding in autumn and spring. The authors provide quite an extensive amount of information on the reproductive success of eggs laid in each season and the possible ecological factors accounting for differences between seasons. Although the presence of interspecific competitors and predators does not seem to account for pond-specific reproductive success, the survival of tadpoles hatching from eggs laid in spring is severely impaired when tadpoles from the autumn cohort have managed to survive. This intraspecific competition takes the form of a “priority” effect where tadpoles from the autumn cohort outcompete the smaller larvae from the spring cohort. Given this strong priority effect, one might tentatively predict that females laying in spring should avoid ponds with tadpoles from the autumn cohort. Surprisingly, however, the authors did not find any evidence for such avoidance, which might indicate strong constraints on the availability of ponds where females might possibly lay. Assuming that each female can indeed lay both in autumn and spring, how is this bimodal phenology maintained? Would not be worthier to allocate all the eggs to the autumn (or the spring) laying season? Eggs laid in autumn and spring have to face different environmental hazards, reducing their hatching success and the probability to produce metamorphs (for instance, tadpoles hatching from eggs laid in autumn have to overwinter which might be a particularly risky phase). Jourdan-Pineau and coworkers addressed this question by adapting a bet-hedging model that was initially developed to investigate the strategy of allocation into seed dormancy of annual plants (Cohen 1966) to the case of the bimodal phenology of the Parsley Frog. By feeding the model with the parameter values obtained from the field surveys, they found that the two breeding strategies (laying in autumn and in spring) can coexist as long as the probability of breeding success in the autumn cohort is between 20% and 80% (the range of values allowing the coexistence of a bimodal phenology shrinking a little bit when considering that frogs can reproduce 5 times during their lifespan instead of three times). This paper provides a very nice illustration of the importance of combining approaches (here field monitoring to gather data that can be used to feed models) to understand the evolution of peculiar breeding strategies. Although future work should attempt to gather individual-based data (in addition to population data), this work shows that spreading the risk can be an adaptive strategy in environments characterized by strong stochastic variation. References Cohen D (1966) Optimizing reproduction in a randomly varying environment. Journal of Theoretical Biology, 12, 119–129. https://doi.org/10.1016/0022-5193(66)90188-3 Jourdan-Pineau H., Crochet P.-A., David P. (2022) Bimodal breeding phenology in the Parsley Frog Pelodytes punctatus as a bet-hedging strategy in an unpredictable environment despite strong priority effects. bioRxiv, 2022.02.24.481784, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.02.24.481784 | Bimodal breeding phenology in the Parsley Frog Pelodytes punctatus as a bet-hedging strategy in an unpredictable environment despite strong priority effects | Helene Jourdan-Pineau, Pierre-Andre Crochet, Patrice David | <p style="text-align: justify;">When environmental conditions are unpredictable, expressing alternative phenotypes spreads the risk of failure, a mixed strategy called bet-hedging. In the southern part of its range, the Parsley Frog <em>Pelodytes ... | | Adaptation, Evolutionary Ecology, Life History | Gabriele Sorci | 2022-02-28 11:53:00 |




