
Latest recommendations

| Id | Title | Authors | Abstract▲ | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
05 Jun 2018

Pleistocene climate change and the formation of regional species poolsRecent assembly of European biogeographic species poolRecommended by Fabien Condamine based on reviews by 3 anonymous reviewersBiodiversity is unevenly distributed over time, space and the tree of life [1]. The fact that regions are richer than others as exemplified by the latitudinal diversity gradient has fascinated biologists as early as the first explorers travelled around the world [2]. Provincialism was one of the first general features of land biotic distributions noted by famous nineteenth century biologists like the phytogeographers J.D. Hooker and A. de Candolle, and the zoogeographers P.L. Sclater and A.R. Wallace [3]. When these explorers travelled among different places, they were struck by the differences in their biotas (e.g. [4]). The limited distributions of distinctive endemic forms suggested a history of local origin and constrained dispersal. Much biogeographic research has been devoted to identifying areas where groups of organisms originated and began their initial diversification [3]. Complementary efforts found evidence of both historical barriers that blocked the exchange of organisms between adjacent regions and historical corridors that allowed dispersal between currently isolated regions. The result has been a division of the Earth into a hierarchy of regions reflecting patterns of faunal and floral similarities (e.g. regions, subregions, provinces). Therefore a first ensuing question is: “how regional species pools have been assembled through time and space?”, which can be followed by a second question: “what are the ecological and evolutionary processes leading to differences in species richness among species pools?”. To address these questions, the study of Calatayud et al. [5] developed and performed an interesting approach relying on phylogenetic data to identify regional and sub-regional pools of European beetles (using the iconic ground beetle genus Carabus). Specifically, they analysed the processes responsible for the assembly of species pools, by comparing the effects of dispersal barriers, niche similarities and phylogenetic history. They found that Europe could be divided in seven modules that group zoogeographically distinct regions with their associated faunas, and identified a transition zone matching the limit of the ice sheets at Last Glacial Maximum (19k years ago). Deviance of species co-occurrences across regions, across sub-regions and within each region was significantly explained, primarily by environmental niche similarity, and secondarily by spatial connectivity, except for northern regions. Interestingly, southern species pools are mostly separated by dispersal barriers, whereas northern species pools are mainly sorted by their environmental niches. Another important finding of Calatayud et al. [5] is that most phylogenetic structuration occurred during the Pleistocene, and they show how extreme recent historical events (Quaternary glaciations) can profoundly modify the composition and structure of geographic species pools, as opposed to studies showing the role of deep-time evolutionary processes. The study of biogeographic assembly of species pools using phylogenies has never been more exciting and promising than today. Catalayud et al. [5] brings a nice study on the importance of Pleistocene glaciations along with geographical barriers and niche-based processes in structuring the regional faunas of European beetles. The successful development of powerful analytical tools in recent years, in conjunction with the rapid and massive increase in the availability of biological data (including molecular phylogenies, fossils, georeferrenced occurrences and ecological traits), will allow us to disentangle complex evolutionary histories. Although we still face important limitations in data availability and methodological shortcomings, the last decade has witnessed an improvement of our understanding of how historical and biotic triggers are intertwined on shaping the Earth’s stupendous biological diversity. I hope that the Catalayud et al.’s approach (and analytical framework) will help movement in that direction, and that it will provide interesting perspectives for future investigations of other regions. Applied to a European beetle radiation, they were able to tease apart the relative contributions of biotic (niche-based processes) versus abiotic (geographic barriers and climate change) factors. References [1] Rosenzweig ML. 1995. Species diversity in space and time. Cambridge: Cambridge University Press. | Pleistocene climate change and the formation of regional species pools | Joaquín Calatayud, Miguel Á. Rodríguez, Rafael Molina-Venegas, María Leo, José Luís Hórreo, Joaquín Hortal | <p>Despite the description of bioregions dates back from the origin of biogeography, the processes originating their associated species pools have been seldom studied. Ancient historical events are thought to play a fundamental role in configuring... | | Phylogeography & Biogeography | Fabien Condamine | 2017-06-14 07:30:32 | ||
07 Aug 2023

Pollen-feeding delays reproductive senescence and maintains toxicity of Heliconius eratoImpact of pollen-feeding on egg-laying and cyanogenic glucoside abundance in red postman butterfliesRecommended by Adriana Briscoe based on reviews by Carol Boggs, Caroline Mueller and 1 anonymous reviewerGrowth, development and reproduction in animals are all limited by dietary nutrients. Expansion of an organism’s diet to sources not accessible to closely related species reduces food competition, and eases the constraints of nutrient-limited diets. Adult butterflies are herbivorous insects known to feed primarily on nectar from flowers, which is rich in sugars but poor in amino acids. Only certain species in the genus Heliconius are known to also feed on pollen, which is especially rich in amino acids, and is known to prolong their lives by several months. The ability to digest pollen in Heliconius has been linked to specialized feeding behaviors (Krenn et al. 2009) and extra-oral digestion using enzymes, possibly including duplicated copies of cocoonase (Harpel et al. 2016; Smith et al. 2016 and 2018), a protease used by some moths to digest silk upon eclosion from their cocoons. In this reprint, Pinheiro de Castro and colleagues investigated the impact of artificial and natural diets on egg-laying ability, body weight, and cyanogenic glucoside abundance in adult Heliconius erato butterflies of both sexes. Previous studies (Dunlap-Pianka et al. 1981) in H. charithonia demonstrated that access to dietary pollen led to extended egg-laying ability among adult female butterflies compared to females deprived of pollen, and compared to Dryas iulia females which feed only on nectar. In the current study, Pinheiro de Castro et al. (2023) examine the impact of diet on both young and old H. erato, over a longer period of time than the earlier work, highlighting the importance of extending the time period over which effects are evaluated. In addition to extending egg-laying ability in older females, the authors found that pollen in the diet appeared to maintain older female body weight, presumably because the pollen contained nutrients depleted during egg-laying. The authors then investigated the effects of nutrition on the production of cyanogenic glycoside defenses. Heliconius are aposematic butterflies that sequester cyanide-forming defense chemicals from food plants as larvae or synthesize these compounds de novo. The authors found the abundance of cyanogenic glycosides to be significantly greater in butterflies with access to pollen, but again only in older females. Curiously, field studies of male and female H. charithonia butterflies found that females in the wild collected more pollen than males (Mendoza-Cuenca and Macías-Ordóñez 2005). Taken together, these new findings raise the intriguing possibility that females collect more pollen than males, in part, because pollen has a bigger impact on female survival and reproduction. A small limitation of the study is the use of wing length, rather than body weight, at the zero time point. But the trend is clear in both males and females, and it adds supporting detail to the efficacy of pollen feeding as an unusual strategy for increasing fertility and survival in Heliconius butterflies.
References | Pollen-feeding delays reproductive senescence and maintains toxicity of Heliconius erato | Erika C. Pinheiro de Castro, Josie McPherson, Glennis Jullian, Anniina L. K. Mattila, Søren Bak, Stephen Montgomery, Chris Jiggins | <p>Dietary shifts may act to ease energetic constraints and allow organisms to optimise life-history traits. Heliconius butterflies differ from other nectar-feeders due to their unique ability to digest pollen, which provides a reliable source of ... | | Evolutionary Ecology, Life History | Adriana Briscoe | 2023-02-07 12:59:54 | ||
07 Sep 2018

Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineagesGenomic parallelism in adaptation to orthogonal environments in sea horsesRecommended by Yaniv Brandvain based on reviews by 2 anonymous reviewersStudies in speciation genomics have revealed that gene flow is quite common, and that despite this, species can maintain their distinct environmental adaptations. Although researchers are still elucidating the genomic mechanisms by which species maintain their adaptations in the face of gene flow, this often appears to involve few diverged genomic regions in otherwise largely undifferentiated genomes. In this preprint [1], Riquet and colleagues investigate the genetic structuring and patterns of parallel evolution in the long-snouted seahorse. References [1] Riquet, F., Liautard-Haag, C., Woodall, L., Bouza, C., Louisy, P., Hamer, B., Otero-Ferrer, F., Aublanc, P., Béduneau, V., Briard, O., El Ayari, T., Hochscheid, S. Belkhir, K., Arnaud-Haond, S., Gagnaire, P.-A., Bierne, N. (2018). Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineages. bioRxiv, 161786, ver. 4 recommended and peer-reviewed by PCI Evol Biol. doi: 10.1101/161786 | Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineages | Florentine Riquet, Cathy Liautard-Haag, Lucy Woodall, Carmen Bouza, Patrick Louisy, Bojan Hamer, Francisco Otero-Ferrer, Philippe Aublanc, Vickie Béduneau, Olivier Briard, Tahani El Ayari, Sandra Hochscheid, Khalid Belkhir, Sophie Arnaud-Haond, Pi... | <p>Diverging semi-isolated lineages either meet in narrow clinal hybrid zones, or have a mosaic distribution associated with environmental variation. Intrinsic reproductive isolation is often emphasized in the former and local adaptation in the la... | | Hybridization / Introgression, Molecular Evolution, Population Genetics / Genomics, Speciation | Yaniv Brandvain | Kathleen Lotterhos, Sarah Fitzpatrick | 2017-07-11 13:12:40 | |
24 Mar 2023
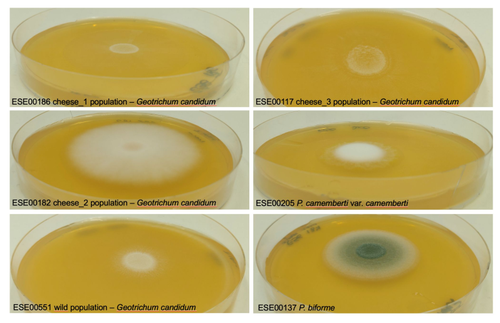
Domestication of different varieties in the cheese-making fungus Geotrichum candidumDiverse outcomes in cheese fungi domesticationRecommended by Christelle Fraïsse based on reviews by Delphine Sicard and 1 anonymous reviewer based on reviews by Delphine Sicard and 1 anonymous reviewer
Domestication is a complex process that imprints the demography and the genomes of domesticated populations, enforcing strong selective pressures on traits favourable to humans, e.g. for food production [1]. Domestication has been quite intensely studied in plants and animals, but less so in micro-organisms such as fungi, despite their assets (e.g. their small genomes and tractability in the lab). This elegant study by Bennetot and collaborators [2] on the cheese-making fungus Geotrichum candidum adds to the mounting body of studies in the genomics of fungi, proving they are excellent models in evolutionary biology for studying adaptation and drift in eukaryotes [3]. Bennetot et al. newly showed with whole genome sequences that all G. candidum strains isolated from cheese form a monophyletic clade subdivided into three genetically differentiated populations with several admixed strains, while the wild strains sampled from diverse geographic locations form a sister clade. This suggests the wild progenitor was not sampled in the present study and calls for future exciting work on the domestication history of the G. candidum fungus. The authors scanned the genomes for footprints of adaptation to the cheese environment and identified promising candidates, such as a gene involved in iron uptake (this element is limiting in cheese). Their functional genome analysis also provides evidence for higher contents of transposable elements in cheese-making strains, likely due to relaxed selection during the domestication process. This paper is particularly impressive in that the authors complemented the population genomic approach with the phenotypic characterization of the strains and tested their ability to outcompete common fungal food spoilers. The authors convincingly showed that cheese-making strains display phenotypic differences relative to wild relatives for multiple traits such as slower growth, lower proteolysis activity and a greater amount of volatiles attractive to consumers, these phenotypes being beneficial for cheese making. Finally, this work is particularly inspiring because it thoroughly discusses convergent evolution during domestication in different cheese-associated fungi. Indeed, studying populations experiencing similar environmental pressures is fundamental to understanding whether evolution is repeatable [4]. For instance, all three cheese populations of G. candidum exhibit a lower genetic diversity than wild populations. However, only one population displays a stronger domestication syndrome, resembling the Penicillium camemberti situation [5]. Furthermore, different cheese-making practices may have led to varying situations with clonal lineages in non-Roquefort P. roqueforti and P. camemberti [5, 6], while the cheese-making G. candidum populations still harbour some diversity. In a nutshell, Bennetot's study makes an important contribution to evolutionary biology and highlights the value of diversifying our model organisms toward under-represented clades. REFERENCES [1] Diamond J (2002) Evolution, consequences and future of plant and animal domestication. Nature 418: 700–707. https://doi.org/10.1038/nature01019 [2] Bennetot B, Vernadet J-P, Perkins V, Hautefeuille S, Rodríguez de la Vega RC, O’Donnell S, Snirc A, Grondin C, Lessard M-H, Peron A-C, Labrie S, Landaud S, Giraud T, Ropars J (2023) Domestication of different varieties in the cheese-making fungus Geotrichum candidum. bioRxiv, 2022.05.17.492043, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.05.17.492043 [3] Gladieux P, Ropars J, Badouin H, Branca A, Aguileta G, de Vienne DM, Rodríguez de la Vega RC, Branco S, Giraud T (2014) Fungal evolutionary genomics provides insight into the mechanisms of adaptive divergence in eukaryotes. Mol. Ecol. 23: 753–773. https://doi.org/10.1111/mec.12631 [4] Bolnick DI, Barrett RD, Oke KB, Rennison DJ, Stuart YE (2018) (Non)Parallel evolution. Ann. Rev. Ecol. Evol. Syst. 49: 303–330. https://doi.org/10.1146/annurev-ecolsys-110617-062240 [5] Ropars J, Didiot E, Rodríguez de la Vega RC, Bennetot B, Coton M, Poirier E, Coton E, Snirc A, Le Prieur S, Giraud T (2020) Domestication of the Emblematic White Cheese-Making Fungus Penicillium camemberti and Its Diversification into Two Varieties. Current Biol. 30: 4441–4453.e4. https://doi.org/10.1016/j.cub.2020.08.082 [6] Dumas, E, Feurtey, A, Rodríguez de la Vega, RC, Le Prieur S, Snirc A, Coton M, Thierry A, Coton E, Le Piver M, Roueyre D, Ropars J, Branca A, Giraud T (2020) Independent domestication events in the blue-cheese fungus Penicillium roqueforti. Mol Ecol. 29: 2639–2660. https://doi.org/10.1111/mec.15359 | Domestication of different varieties in the cheese-making fungus *Geotrichum candidum* | Bastien Bennetot, Jean-Philippe Vernadet, Vincent Perkins, Sophie Hautefeuille, Ricardo C. Rodríguez de la Vega, Samuel O’Donnell, Alodie Snirc, Cécile Grondin, Marie-Hélène Lessard, Anne-Claire Peron, Steve Labrie, Sophie Landaud, Tatiana Giraud,... | <p>Domestication is an excellent model for studying adaptation processes, involving recent adaptation and diversification, convergence following adaptation to similar conditions, as well as degeneration of unused functions. <em>Geotrichum candidum... | | Adaptation, Genome Evolution, Population Genetics / Genomics | Christelle Fraïsse | 2022-08-12 20:50:42 | ||
19 Feb 2018

Genomic imprinting mediates dosage compensation in a young plant XY systemDosage compensation by upregulation of maternal X alleles in both males and females in young plant sex chromosomesRecommended by Tatiana Giraud and Judith Mank based on reviews by 3 anonymous reviewersSex chromosomes evolve as recombination is suppressed between the X and Y chromosomes. The loss of recombination on the sex-limited chromosome (the Y in mammals) leads to degeneration of both gene expression and gene content for many genes [1]. Loss of gene expression or content from the Y chromosome leads to differences in gene dose between males and females for X-linked genes. Because expression levels are often correlated with gene dose [2], these hemizygous genes have a lower expression levels in the heterogametic sex. This in turn disrupts the stoichiometric balance among genes in protein complexes that have components on both the sex chromosomes and autosomes [3], which could have serious deleterious consequences for the heterogametic sex. References | Genomic imprinting mediates dosage compensation in a young plant XY system | Aline Muyle, Niklaus Zemp, Cecile Fruchard, Radim Cegan, Jan Vrana, Clothilde Deschamps, Raquel Tavares, Franck Picard, Roman Hobza, Alex Widmer, Gabriel Marais | <p>During the evolution of sex chromosomes, the Y degenerates and its expression gets reduced relative to the X and autosomes. Various dosage compensation mechanisms that recover ancestral expression levels in males have been described in animals.... | | Bioinformatics & Computational Biology, Expression Studies, Genome Evolution, Molecular Evolution, Reproduction and Sex | Tatiana Giraud | 2017-09-20 20:39:46 | ||
21 Nov 2019
Environmental specificity in Drosophila-bacteria symbiosis affects host developmental plasticityNutrition-dependent effects of gut bacteria on growth plasticity in Drosophila melanogasterRecommended by Wolf Blanckenhorn based on reviews by Pedro Simões and 1 anonymous reviewerIt is well known that the rearing environment has strong effects on life history and fitness traits of organisms. Microbes are part of every environment and as such likely contribute to such environmental effects. Gut bacteria are a special type of microbe that most animals harbor, and as such they are part of most animals’ environment. Such microbial symbionts therefore likely contribute to local adaptation [1]. The main question underlying the laboratory study by Guilhot et al. [2] was: How much do particular gut bacteria affect the organismal phenotype, in terms of life history and larval foraging traits, of the fruit fly Drosophila melanogaster, a common laboratory model species in biology? References [1] Kawecki, T. J. and Ebert, D. (2004) Conceptual issues in local adaptation. Ecology Letters 7: 1225-1241. doi: 10.1111/j.1461-0248.2004.00684.x | Environmental specificity in Drosophila-bacteria symbiosis affects host developmental plasticity | Robin Guilhot, Antoine Rombaut, Anne Xuéreb, Kate Howell, Simon Fellous | <p>Environmentally acquired microbial symbionts could contribute to host adaptation to local conditions like vertically transmitted symbionts do. This scenario necessitates symbionts to have different effects in different environments. We investig... | | Adaptation, Evolutionary Ecology, Phenotypic Plasticity, Species interactions | Wolf Blanckenhorn | 2019-02-13 15:22:23 | ||
02 Nov 2020

Experimental evolution of virulence and associated traits in a Drosophila melanogaster – Wolbachia symbiosisTemperature effects on virulence evolution of wMelPop Wolbachia in Drosophila melanogasterRecommended by Ellen Decaestecker based on reviews by Shira Houwenhuyse and 3 anonymous reviewersMonnin et al. [1] here studied how Drosophila populations are affected when exposed to a high virulent endosymbiotic wMelPop Wolbachia strain and why virulent vertically transmitting endosymbionts persist in nature. This virulent wMelPop strain has been described to be a blocker of dengue and other arboviral infections in arthropod vector species, such as Aedes aegypti. Whereas it can thus function as a mutualistic symbiont, it here acts as an antagonist along the mutualism-antagonism continuum symbionts operate. The wMelPop strain is not a natural occurring strain in Drosophila melanogaster and thus the start of this experiment can be seen as a novel host-pathogen association. Through experimental evolution of 17 generations, the authors studied how high temperature affects wMelPop Wolbachia virulence and Drosophila melanogaster survival. The authors used Drosophila strains that were selected for late reproduction, given that this should favor evolution to a lower virulence. Assumptions for this hypothesis are not given in the manuscript here, but it can indeed be assumed that energy that is assimilated to symbiont tolerance instead of reproduction may lead to reduced virulence evolution. This has equally been suggested by Reyserhove et al. [2] in a dynamics energy budget model tailored to Daphnia magna virulence evolution upon a viral infection causing White fat Cell disease, reconstructing changing environments through time. References [1] Monnin, D., Kremer, N., Michaud, C., Villa, M., Henri, H., Desouhant, E. and Vavre, F. (2020) Experimental evolution of virulence and associated traits in a Drosophila melanogaster – Wolbachia symbiosis. bioRxiv, 2020.04.26.062265, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: https://doi.org/10.1101/2020.04.26.062265 | Experimental evolution of virulence and associated traits in a Drosophila melanogaster – Wolbachia symbiosis | David Monnin, Natacha Kremer, Caroline Michaud, Manon Villa, Hélène Henri, Emmanuel Desouhant, Fabrice Vavre | <p>Evolutionary theory predicts that vertically transmitted symbionts are selected for low virulence, as their fitness is directly correlated to that of their host. In contrast with this prediction, the Wolbachia strain wMelPop drastically reduces... | | Evolutionary Ecology, Experimental Evolution, Species interactions | Ellen Decaestecker | 2020-04-29 19:16:56 | ||
03 May 2020

When does gene flow facilitate evolutionary rescue?Reconciling the upsides and downsides of migration for evolutionary rescueRecommended by Claudia Bank based on reviews by 3 anonymous reviewersThe evolutionary response of populations to changing or novel environments is a topic that unites the interests of evolutionary biologists, ecologists, and biomedical researchers [1]. A prominent phenomenon in this research area is evolutionary rescue, whereby a population that is otherwise doomed to extinction survives due to the spread of new or pre-existing mutations that are beneficial in the new environment. Scenarios of evolutionary rescue require a specific set of parameters: the absolute growth rate has to be negative before the rescue mechanism spreads, upon which the growth rate becomes positive. However, potential examples of its relevance exist (e.g., [2]). From a theoretical point of view, the technical challenge but also the beauty of evolutionary rescue models is that they combine the study of population dynamics (i.e., changes in the size of populations) and population genetics (i.e., changes in the frequencies in the population). Together, the potential relevance of evolutionary rescue in nature and the models' theoretical appeal has resulted in a suite of modeling studies on the subject in recent years. References [1] Bell, G. (2017). Evolutionary Rescue. Annual Review of Ecology, Evolution, and Systematics 48(1), 605-627. doi: 10.1146/annurev-ecolsys-110316-023011 | When does gene flow facilitate evolutionary rescue? | Matteo Tomasini, Stephan Peischl | <p>Experimental and theoretical studies have highlighted the impact of gene flow on the probability of evolutionary rescue in structured habitats. Mathematical modelling and simulations of evolutionary rescue in spatially or otherwise structured p... | | Evolutionary Dynamics, Evolutionary Theory, Population Genetics / Genomics | Claudia Bank | 2019-05-22 11:12:13 | ||
23 Apr 2020

How do invasion syndromes evolve? An experimental evolution approach using the ladybird Harmonia axyridisSelection on a single trait does not recapitulate the evolution of life-history traits seen during an invasionRecommended by Inês Fragata and Ben Phillips based on reviews by 2 anonymous reviewersBiological invasions are natural experiments, and often show that evolution can affect dynamics in important ways [1-3]. While we often think of invasions as a conservation problem stemming from anthropogenic introductions [4,5], biological invasions are much more commonplace than this, including phenomena as diverse as natural range shifts, the spread of novel pathogens, and the growth of tumors. A major question across all these settings is which set of traits determine the ability of a population to invade new space [6,7]. Traits such as: increased growth or reproductive rate, dispersal ability and ability to defend from predation often show large evolutionary shifts across invasion history [1,6,8]. Are such multi-trait shifts driven by selection on multiple traits, or a correlated response by multiple traits to selection on one? Resolving this question is important for both theoretical and practical reasons [9,10]. But despite the importance of this issue, it is not easy to perform the necessary manipulative experiments [9]. References [1] Sakai, A.K., Allendorf, F.W., Holt, J.S. et al. (2001). The population biology of invasive species. Annual review of ecology and systematics, 32(1), 305-332. doi: 10.1146/annurev.ecolsys.32.081501.114037 | How do invasion syndromes evolve? An experimental evolution approach using the ladybird Harmonia axyridis | Julien Foucaud, Ruth A. Hufbauer, Virginie Ravigné, Laure Olazcuaga, Anne Loiseau, Aurelien Ausset, Su Wang, Lian-Sheng Zang, Nicolas Lemenager, Ashraf Tayeh, Arthur Weyna, Pauline Gneux, Elise Bonnet, Vincent Dreuilhe, Bastien Poutout, Arnaud Est... | <p>Experiments comparing native to introduced populations or distinct introduced populations to each other show that phenotypic evolution is common and often involves a suit of interacting phenotypic traits. We define such sets of traits that evol... | | Adaptation, Evolutionary Applications, Experimental Evolution, Life History, Quantitative Genetics | Inês Fragata | 2019-11-29 07:07:00 | ||
03 Oct 2018
Range size dynamics can explain why evolutionarily age and diversification rate correlate with contemporary extinction risk in plantsAre both very young and the very old plant lineages at heightened risk of extinction?Recommended by Arne Mooers based on reviews by Dan Greenberg and 1 anonymous reviewerHuman economic activity is responsible for the vast majority of ongoing extinction, but that does not mean lineages are being affected willy-nilly. For amphibians [1] and South African flowering plants [2], young species have a somewhat higher than expected chance of being threatened with extinction. In contrast, older Australian marsupial lineages seem to be more at risk [3]. Both of the former studies suggested that situations leading to peripheral isolation might simultaneously increase ongoing speciation and current threat via small geographic range, while the authors of the latter study suggested that older species might have evolved increasingly narrow niches. Here, Andrew Tanentzap and colleagues [4] dig deeper into the putative links between species age, niche breadth and threat status. Across 500-some plant genera worldwide, they find that, indeed, ""younger"" species (i.e. from younger and faster-diversifying genera) were more likely to be listed as imperiled by the IUCN, consistent with patterns for amphibians and African plants. Given this, results from their finer-level analyses of conifers are initially bemusing: here, ""older"" (i.e., on longer terminal branches) species were at higher risk. This would make conifers more like Australian marsupials, with the rest of the plants being more like amphibians. However, here where the data were more finely grained, the authors detected a second interesting pattern: using an intriguing matched-pair design, they detect a signal of conifer species niches seemingly shrinking as a function of age. The authors interpret this as consistent with increasing specialization, or loss of ancestral warm wet habitat, over paleontological time. It is true that conifers in general are older than plants more generally, with some species on branches that extend back many 10s of millions of years, and so a general loss of suitable habitat makes some sense. If so, both the pattern for all plants (small initial ranges heightening extinction) and the pattern for conifers (eventual increasing specialization or habitat contraction heightening extinction) could occur, each on a different time scale. As a coda, the authors detected no effect of age on threat status in palms; however, this may be both because palms have already lost species to climate-change induced extinction, and because they are thought to speciate more via long-distance dispersal and adaptive divergence then via peripheral isolation. References [1] Greenberg, D. A., & Mooers, A. Ø. (2017). Linking speciation to extinction: Diversification raises contemporary extinction risk in amphibians. Evolution Letters, 1, 40–48. doi: 10.1002/evl3.4 | Range size dynamics can explain why evolutionarily age and diversification rate correlate with contemporary extinction risk in plants | Andrew J. Tanentzap, Javier Igea, Matthew G. Johnston, Matthew J. Larcombe | <p>Extinction threatens many species, yet few factors predict this risk across the plant Tree of Life (ToL). Taxon age is one factor that may associate with extinction if occupancy of geographic and adaptive zones varies with time, but evidence fo... | | Macroevolution, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Arne Mooers | 2018-02-01 21:01:19 |




