
Latest recommendations

| Id | Title | Authors▲ | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
10 Jan 2019

Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruceDisentangling the recent and ancient demographic history of European spruce speciesRecommended by Jason Holliday based on reviews by 1 anonymous reviewerGenetic diversity in temperate and boreal forests tree species has been strongly affected by late Pleistocene climate oscillations [2,3,5], but also by anthropogenic forces. Particularly in Europe, where a long history of human intervention has re-distributed species and populations, it can be difficult to know if a given forest arose through natural regeneration and gene flow or through some combination of natural and human-mediated processes. This uncertainty can confound inferences of the causes and consequences of standing genetic variation, which may impact our interpretation of demographic events that shaped species before humans became dominant on the landscape. In their paper entitled "Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce", Chen et al. [1] used a genome-wide dataset of 400k SNPs to infer the demographic history of Picea abies (Norway spruce), the most widespread and abundant spruce species in Europe, and to understand its evolutionary relationship with two other spruces (Picea obovata [Siberian spruce] and P. omorika [Serbian spruce]). Three major Norway spruce clusters were identified, corresponding to central Europe, Russia and the Baltics, and Scandinavia, which agrees with previous studies. The density of the SNP data in the present paper enabled inference of previously uncharacterized admixture between these groups, which corresponds to the timing of postglacial recolonization following the last glacial maximum (LGM). This suggests that multiple migration routes gave rise to the extant distribution of the species, and may explain why Chen et al.'s estimates of divergence times among these major Norway spruce groups were older (15mya) than those of previous studies (5-6mya) – those previous studies may have unknowingly included admixed material [4]. Treemix analysis also revealed extensive admixture between Norway and Siberian spruce over the last ~100k years, while the geographically-restricted Serbian spruce was both isolated from introgression and had a dramatically smaller effective population size (Ne) than either of the other two species. This small Ne resulted from a bottleneck associated with the onset of the iron age ~3000 years ago, which suggests that anthropogenic depletion of forest resources has severely impacted this species. Finally, ancestry of Norway spruce samples collected in Sweden and Denmark suggest their recent transfer from more southern areas of the species range. This northward movement of genotypes likely occurred because the trees performed well relative to local provenances, which is a common observation when trees from the south are planted in more northern locations (although at the potential cost of frost damage due to inappropriate phenology). While not the reason for the transfer, the incorporation of southern seed sources into the Swedish breeding and reforestation program may lead to more resilient forests under climate change. Taken together, the data and analysis presented in this paper allowed inference of the intra- and interspecific demographic histories of a tree species group at a very high resolution, and suggest caveats regarding sampling and interpretation of data from areas with a long history of occupancy by humans. References [1] Chen, J., Milesi, P., Jansson, G., Berlin, M., Karlsson, B., Aleksić, J. M., Vendramin, G. G., Lascoux, M. (2018). Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce. BioRxiv, 402016. ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/402016 | Genomic data provides new insights on the demographic history and the extent of recent material transfers in Norway spruce | Jun Chen, Lili Li, Pascal Milesi, Gunnar Jansson, Mats Berlin, Bo Karlsson, Jelena Aleksic, Giovanni G Vendramin, Martin Lascoux | <p>Primeval forests are today exceedingly rare in Europe and transfer of forest reproductive material for afforestation and improvement have been very common, especially over the last two centuries. This can be a serious impediment when inferring ... | | Evolutionary Applications, Hybridization / Introgression, Population Genetics / Genomics | Jason Holliday | Anonymous, Anonymous | 2018-08-29 08:33:15 | |
14 Feb 2024
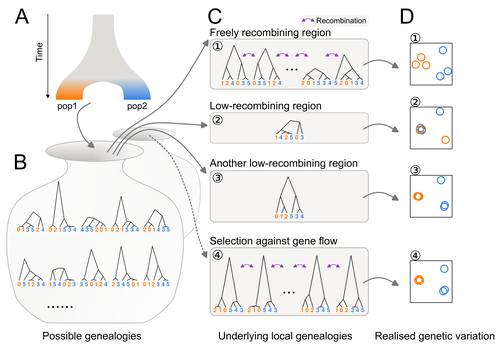
Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structureDiscerning the causes of local deviations in genetic variation: the effect of low-recombination regionsRecommended by Matteo Fumagalli based on reviews by Claire Merot and 1 anonymous reviewer based on reviews by Claire Merot and 1 anonymous reviewer
In this study, Ishigohoka and colleagues tackle an important, yet often overlooked, question on the causes of genetic variation. While genome-wide patterns represent population structure, local variation is often associated with selection. Authors propose that an alternative cause for variation in individual loci is reduced recombination rate. To test this hypothesis, authors perform local Principal Component Analysis (PCA) (Li & Ralph, 2019) to identify local deviations in population structure in the Eurasian blackcap (Sylvia atricapilla) (Ishigohoka et al. 2022). This approach is typically used to detect chromosomal rearrangements or any long region of linked loci (e.g., due to reduced recombination or selection) (Mérot et al. 2021). While other studies investigated the effect of low recombination on genetic variation (Booker et al. 2020), here authors provide a comprehensive analysis of the effect of recombination to local PCA patterns both in empirical and simulated data sets. Findings demonstrate that low recombination (and not selection) can be the sole explanatory variable for outlier windows. The study also describes patterns of genetic variation along the genome of Eurasian blackcaps, localising at least two polymorphic inversions (Ishigohoka et al. 2022). Further investigations on the effect of model parameters (e.g., window sizes and thresholds for defining low-recombining regions), as well as the use of powerful neutrality tests are in need to clearly assess whether outlier regions experience selection and reduced recombination, and to what extent. References Booker, T. R., Yeaman, S., & Whitlock, M. C. (2020). Variation in recombination rate affects detection of outliers in genome scans under neutrality. Molecular Ecology, 29 (22), 4274–4279. https://doi.org/10.1111/mec.15501 Ishigohoka, J., Bascón-Cardozo, K., Bours, A., Fuß, J., Rhie, A., Mountcastle, J., Haase, B., Chow, W., Collins, J., Howe, K., Uliano-Silva, M., Fedrigo, O., Jarvis, E. D., Pérez-Tris, J., Illera, J. C., Liedvogel, M. (2022) Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structure. bioRxiv 2021.12.22.473882, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.12.22.473882 Li, H., & Ralph, P. (2019). Local PCA Shows How the Effect of Population Structure Differs Along the Genome. Genetics, 211 (1), 289–304. https://doi.org/10.1534/genetics.118.301747 Mérot, C., Berdan, E. L., Cayuela, H., Djambazian, H., Ferchaud, A.-L., Laporte, M., Normandeau, E., Ragoussis, J., Wellenreuther, M., & Bernatchez, L. (2021). Locally Adaptive Inversions Modulate Genetic Variation at Different Geographic Scales in a Seaweed Fly. Molecular Biology and Evolution, 38 (9), 3953–3971. https://doi.org/10.1093/molbev/msab143 | Distinct patterns of genetic variation at low-recombining genomic regions represent haplotype structure | Jun Ishigohoka, Karen Bascón-Cardozo, Andrea Bours, Janina Fuß, Arang Rhie, Jacquelyn Mountcastle, Bettina Haase, William Chow, Joanna Collins, Kerstin Howe, Marcela Uliano-Silva, Olivier Fedrigo, Erich D. Jarvis, Javier Pérez-Tris, Juan Carlos Il... | <p>Genetic variation of the entire genome represents population structure, yet individual loci can show distinct patterns. Such deviations identified through genome scans have often been attributed to effects of selection instead of randomness. Th... | | Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Matteo Fumagalli | 2023-10-13 11:58:47 | ||
01 Jul 2022

Genomic evidence of paternal genome elimination in the globular springtail Allacma fuscaPressing NGS data through the mill of Kmer spectra and allelic coverage ratios in order to scan reproductive modes in non-model speciesRecommended by Nicolas Bierne based on reviews by Paul Simion and 2 anonymous reviewersThe genomic revolution has given us access to inexpensive genetic data for any species. Simultaneously we have lost the ability to easily identify chimerism in samples or some unusual deviations from standard Mendelian genetics. Methods have been developed to identify sex chromosomes, characterise the ploidy, or understand the exact form of parthenogenesis from genomic data. However, we rarely consider that the tissues we extract DNA from could be a mixture of cells with different genotypes or karyotypes. This can nonetheless happen for a variety of (fascinating) reasons such as somatic chromosome elimination, transmissible cancer, or parental genome elimination. Without a dedicated analysis, it is very easy to miss it. In this preprint, Jaron et al. (2022) used an ingenious analysis of whole individual NGS data to test the hypothesis of paternal genome elimination in the globular springtail Allacma fusca. The authors suspected that a high fraction of the whole body of males is made of sperm in this species and if this species undergoes paternal genome elimination, we would expect that sperm would only contain maternally inherited chromosomes. Given the reference genome was highly fragmented, they developed a two-tissue model to analyse Kmer spectra and obtained confirmation that around one-third of the tissue was sperm in males. This allowed them to test whether coverage patterns were consistent with the species exhibiting paternal genome elimination. They combined their estimation of the fraction of haploid tissue with allele coverages in autosomes and the X chromosome to obtain support for a bias toward one parental allele, suggesting that all sperm carries the same parental haplotype. It could be the maternal or the paternal alleles, but paternal genome elimination is most compatible with the known biology of Arthropods. SNP calling was used to confirm conclusions based on the analysis of the raw pileups. I found this study to be a good example of how a clever analysis of Kmer spectra and allele coverages can provide information about unusual modes of reproduction in a species, even though it does not have a well-assembled genome yet. As advocated by the authors, routine inspection of Kmer spectra and allelic read-count distributions should be included in the best practice of NGS data analysis. They provide the method to identify paternal genome elimination but also the way to develop similar methods to detect another kind of genetic chimerism in the avalanche of sequence data produced nowadays. References Jaron KS, Hodson CN, Ellers J, Baird SJ, Ross L (2022) Genomic evidence of paternal genome elimination in the globular springtail Allacma fusca. bioRxiv, 2021.11.12.468426, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.11.12.468426 | Genomic evidence of paternal genome elimination in the globular springtail Allacma fusca | Kamil S. Jaron, Christina N. Hodson, Jacintha Ellers, Stuart JE Baird, Laura Ross | <p style="text-align: justify;">Paternal genome elimination (PGE) - a type of reproduction in which males inherit but fail to pass on their father’s genome - evolved independently in six to eight arthropod clades. Thousands of species, including s... | | Genome Evolution, Reproduction and Sex | Nicolas Bierne | 2021-11-18 00:09:43 | ||
02 Feb 2023
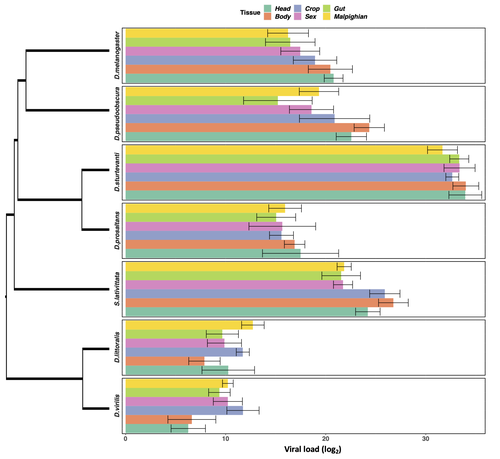
Heterogeneities in infection outcomes across species: sex and tissue differences in virus susceptibilitySusceptibility to infection is not explained by sex or differences in tissue tropism across different species of DrosophilaRecommended by Alison Duncan based on reviews by Greg Hurst and 1 anonymous reviewerUnderstanding factors explaining both intra and interspecific variation in susceptibility to infection by parasites remains a key question in evolutionary biology. Within a species variation in susceptibility is often explained by differences in behaviour affecting exposure to infection and/or resistance affecting the degree by which parasite growth is controlled (Roy & Kirchner, 2000, Behringer et al., 2000). This can vary between the sexes (Kelly et al., 2018) and may be explained by the ability of a parasite to attack different organs or tissues (Brierley et al., 2019). However, what goes on within one species is not always relevant to another, making it unclear when patterns can be scaled up and generalised across species. This is also important to understand when parasites may jump hosts, or identify species that may be susceptible to a host jump (Longdon et al., 2015). Phylogenetic distance between hosts is often an important factor explaining susceptibility to a particular parasite in plant and animal hosts (Gilbert & Webb, 2007, Faria et al., 2013). In two separate experiments, Roberts and Longdon (Roberts & Longdon, 2022) investigated how sex and tissue tropism affected variation in the load of Drosophila C Virus (DCV) across multiple Drosophila species. DCV load has been shown to correlate positively with mortality (Longdon et al., 2015). Overall, they found that load did not vary between the sexes; within a species males and females had similar DCV loads for 31 different species. There was some variation in levels of DCV growth in different tissue types, but these too were consistent across males for 7 species of Drosophila. Instead, in both experiments, host phylogeny or interspecific variation, explained differences in DCV load with some species being more infected than others. This study is neat in that it incorporates and explores simultaneously both intra and interspecific variation in infection-related life-history traits which is not often done (but see (Longdon et al., 2015, Imrie et al., 2021, Longdon et al., 2011, Johnson et al., 2012). Indeed, most studies to date explore either inter-specific differences in susceptibility to a parasite (it can or can’t infect a given species) (Davies & Pedersen, 2008, Pfenning-Butterworth et al., 2021) or intra-specific variability in infection-related traits (infectivity, resistance etc.) due to factors such as sex, genotype and environment (Vale et al., 2008, Lambrechts et al., 2006). This work thus advances on previous studies, while at the same time showing that sex differences in parasite load are not necessarily pervasive. References Behringer DC, Butler MJ, Shields JD (2006) Avoidance of disease by social lobsters. Nature, 441, 421–421. https://doi.org/10.1038/441421a Brierley L, Pedersen AB, Woolhouse MEJ (2019) Tissue tropism and transmission ecology predict virulence of human RNA viruses. PLOS Biology, 17, e3000206. https://doi.org/10.1371/journal.pbio.3000206 Davies TJ, Pedersen AB (2008) Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proceedings of the Royal Society B: Biological Sciences, 275, 1695–1701. https://doi.org/10.1098/rspb.2008.0284 Faria NR, Suchard MA, Rambaut A, Streicker DG, Lemey P (2013) Simultaneously reconstructing viral cross-species transmission history and identifying the underlying constraints. Philosophical Transactions of the Royal Society B: Biological Sciences, 368, 20120196. https://doi.org/10.1098/rstb.2012.0196 Gilbert GS, Webb CO (2007) Phylogenetic signal in plant pathogen–host range. Proceedings of the National Academy of Sciences, 104, 4979–4983. https://doi.org/10.1073/pnas.0607968104 Imrie RM, Roberts KE, Longdon B (2021) Between virus correlations in the outcome of infection across host species: Evidence of virus by host species interactions. Evolution Letters, 5, 472–483. https://doi.org/10.1002/evl3.247 Johnson PTJ, Rohr JR, Hoverman JT, Kellermanns E, Bowerman J, Lunde KB (2012) Living fast and dying of infection: host life history drives interspecific variation in infection and disease risk. Ecology Letters, 15, 235–242. https://doi.org/10.1111/j.1461-0248.2011.01730.x Kelly CD, Stoehr AM, Nunn C, Smyth KN, Prokop ZM (2018) Sexual dimorphism in immunity across animals: a meta-analysis. Ecology Letters, 21, 1885–1894. https://doi.org/10.1111/ele.13164 Lambrechts L, Chavatte J-M, Snounou G, Koella JC (2006) Environmental influence on the genetic basis of mosquito resistance to malaria parasites. Proceedings of the Royal Society B: Biological Sciences, 273, 1501–1506. https://doi.org/10.1098/rspb.2006.3483 Longdon B, Hadfield JD, Day JP, Smith SCL, McGonigle JE, Cogni R, Cao C, Jiggins FM (2015) The Causes and Consequences of Changes in Virulence following Pathogen Host Shifts. PLOS Pathogens, 11, e1004728. https://doi.org/10.1371/journal.ppat.1004728 Longdon B, Hadfield JD, Webster CL, Obbard DJ, Jiggins FM (2011) Host Phylogeny Determines Viral Persistence and Replication in Novel Hosts. PLOS Pathogens, 7, e1002260. https://doi.org/10.1371/journal.ppat.1002260 Pfenning-Butterworth AC, Davies TJ, Cressler CE (2021) Identifying co-phylogenetic hotspots for zoonotic disease. Philosophical Transactions of the Royal Society B: Biological Sciences, 376, 20200363. https://doi.org/10.1098/rstb.2020.0363 Roberts KE, Longdon B (2023) Heterogeneities in infection outcomes across species: examining sex and tissue differences in virus susceptibility. bioRxiv 2022.11.01.514663, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.11.01.514663 Roy BA, Kirchner JW (2000) Evolutionary Dynamics of Pathogen Resistance and Tolerance. Evolution, 54, 51–63. https://doi.org/10.1111/j.0014-3820.2000.tb00007.x Vale PF, Stjernman M, Little TJ (2008) Temperature-dependent costs of parasitism and maintenance of polymorphism under genotype-by-environment interactions. Journal of Evolutionary Biology, 21, 1418–1427. https://doi.org/10.1111/j.1420-9101.2008.01555.x | Heterogeneities in infection outcomes across species: sex and tissue differences in virus susceptibility | Katherine E Roberts, Ben Longdon | <p style="text-align: justify;">Species vary in their susceptibility to pathogens, and this can alter the ability of a pathogen to infect a novel host. However, many factors can generate heterogeneity in infection outcomes, obscuring our ability t... | | Evolutionary Ecology | Alison Duncan | Anonymous, Greg Hurst | 2022-11-03 11:17:42 | |
18 May 2018

Modularity of genes involved in local adaptation to climate despite physical linkageDifferential effect of genes in diverse environments, their role in local adaptation and the interference between genes that are physically linkedRecommended by Sebastian Ernesto Ramos-Onsins based on reviews by Tanja Pyhäjärvi and 1 anonymous reviewerThe genome of eukaryotic species is a complex structure that experience many different interactions within itself and with the surrounding environment. The genetic architecture of a phenotype (that is, the set of genetic elements affecting a trait of the organism) plays a fundamental role in understanding the adaptation process of a species to, for example, different climate environments, or to its interaction with other species. Thus, it is fundamental to study the different aspects of the genetic architecture of the species and its relationship with its surronding environment. Aspects such as modularity (the number of genetic units and the degree to which each unit is affecting a trait of the organism), pleiotropy (the number of different effects that a genetic unit can have on an organism) or linkage (the degree of association between the different genetic units) are essential to understand the genetic architecture and to interpret the effects of selection on the genome. Indeed, the knowledge of the different aspects of the genetic architecture could clarify whether genes are affected by multiple aspects of the environment or, on the contrary, are affected by only specific aspects [1,2]. The work performed by Lotterhos et al. [3] sought to understand the genetic architecture of the adaptation to different environments in lodgepole pine (Pinus contorta), considering as candidate SNPs those previously detected as a result of its extreme association patterns to different environmental variables or to extreme population differentiation. This consideration is very important because the study is only relevant if the studied markers are under the effect of selection. Otherwise, the genetic architecture of the adaptation to different environments would be masked by other (neutral) kind of associations that would be difficult to interpret [4,5]. In order to understand the relationship between genetic architecture and adaptation, it is relevant to detect the association networks of the candidate SNPs with climate variables (a way to measure modularity) and if these SNPs (and loci) are affected by single or multiple environments (a way to measure pleiotropy). The authors used co-association networks, an innovative approach in this field, to analyse the interaction between the environmental information and the genetic polymorphism of each individual. This methodology is more appropriate than other multivariate methods - such as analysis based on principal components - because it is possible to cluster SNPs based on associations with similar environmental variables. In this sense, the co-association networks allowed to both study the genetic and physical linkage between different co-associations modules but also to compare two different models of evolution: a Modular environmental response architecture (specific genes are affected by specific aspects of the environment) or a Universal pleiotropic environmental response architecture (all genes are affected by all aspects of the environment). The representation of different correlations between allelic frequency and environmental factors (named galaxy biplots) are especially informative to understand the effect of the different clusters on specific aspects of the environment (for example, the co-association network ‘Aridity’ shows strong associations with hot/wet versus cold/dry environments). The analysis performed by Lotterhos et al. [3], although it has some unavoidable limitations (e.g., only extreme candidate SNPs are selected, limiting the results to the stronger effects; the genetic and physical map is incomplete in this species), includes relevant results and also implements new methodologies in the field. To highlight some of them: the preponderance of a Modular environmental response architecture (evolution in separated modules), the detection of physical linkage among SNPs that are co-associated with different aspects of the environment (which was unexpected a priori), the implementation of co-association networks and galaxy biplots to see the effect of modularity and pleiotropy on different aspects of environment. Finally, this work contains remarkable introductory Figures and Tables explaining unambiguously the main concepts [6] included in this study. This work can be treated as a starting point for many other future studies in the field. References [1] Hancock AM, Brachi B, Faure N, Horton MW, Jarymowycz LB, Sperone FG, Toomajian C, Roux F & Bergelson J. 2011. Adaptation to climate across the Arabidopsis thaliana genome. Science 334: 83–86. doi: 10.1126/science.1209244 | Modularity of genes involved in local adaptation to climate despite physical linkage | Katie E. Lotterhos, Sam Yeaman, Jon Degner, Sally Aitken, Kathryn Hodgins | <p>Background: Physical linkage among genes shaped by different sources of selection is a fundamental aspect of genetic architecture. Theory predicts that evolution in complex environments selects for modular genetic architectures and high recombi... | | Adaptation, Bioinformatics & Computational Biology, Genome Evolution | Sebastian Ernesto Ramos-Onsins | 2017-10-15 19:21:57 | ||
22 May 2023
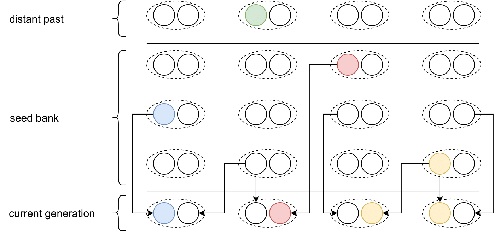
Weak seed banks influence the signature and detectability of selective sweepsNew insights into the dynamics of selective sweeps in seed-banked speciesRecommended by Renaud Vitalis based on reviews by Guillaume Achaz, Jere Koskela, William Shoemaker and Simon Boitard
Many organisms across the Tree of life have the ability to produce seeds, eggs, cysts, or spores, that can remain dormant for several generations before hatching. This widespread adaptive trait in bacteria, fungi, plants and animals, has a significant impact on the ecology, population dynamics and population genetics of species that express it (Evans and Dennehy 2005). In population genetics, and despite the recognition of its evolutionary importance in many empirical studies, few theoretical models have been developed to characterize the evolutionary consequences of this trait on the level and distribution of neutral genetic diversity (see, e.g., Kaj et al. 2001; Vitalis et al. 2004), and also on the dynamics of selected alleles (see, e.g., Živković and Tellier 2018). However, due to the complexity of the interactions between evolutionary forces in the presence of dormancy, the fate of selected mutations in their genomic environment is not yet fully understood, even from the most recently developed models. In a comprehensive article, Korfmann et al. (2023) aim to fill this gap by investigating the effect of germ banking on the probability of (and time to) fixation of beneficial mutations, as well as on the shape of the selective sweep in their vicinity. To this end, Korfmann et al. (2023) developed and released their own forward-in-time simulator of genome-wide data, including neutral and selected polymorphisms, that makes use of Kelleher et al.’s (2018) tree sequence toolkit to keep track of gene genealogies. The originality of Korfmann et al.’s (2023) study is to provide new quantitative results for the effect of dormancy on the time to fixation of positively selected mutations, the shape of the genomic landscape in the vicinity of these mutations, and the temporal dynamics of selective sweeps. Their major finding is the prediction that germ banking creates narrower signatures of sweeps around positively selected sites, which are detectable for increased periods of time (as compared to a standard Wright-Fisher population). The availability of Korfmann et al.’s (2023) code will allow a wider range of parameter values to be explored, to extend their results to the particularities of the biology of many species. However, as they chose to extend the haploid coalescent model of Kaj et al. (2001), further development is needed to confirm the robustness of their results with a more realistic diploid model of seed dormancy. REFERENCES Evans, M. E. K., and J. J. Dennehy (2005) Germ banking: bet-hedging and variable release from egg and seed dormancy. The Quarterly Review of Biology, 80(4): 431-451. https://doi.org/10.1086/498282 Kaj, I., S. Krone, and M. Lascoux (2001) Coalescent theory for seed bank models. Journal of Applied Probability, 38(2): 285-300. https://doi.org/10.1239/jap/996986745 Kelleher, J., K. R. Thornton, J. Ashander, and P. L. Ralph (2018) Efficient pedigree recording for fast population genetics simulation. PLoS Computational Biology, 14(11): e1006581. https://doi.org/10.1371/journal.pcbi.1006581 Korfmann, K., D. Abu Awad, and A. Tellier (2023) Weak seed banks influence the signature and detectability of selective sweeps. bioRxiv, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.04.26.489499 Vitalis, R., S. Glémin, and I. Olivieri (2004) When genes go to sleep: the population genetic consequences of seed dormancy and monocarpic perenniality. American Naturalist, 163(2): 295-311. https://doi.org/10.1086/381041 Živković, D., and A. Tellier (2018). All but sleeping? Consequences of soil seed banks on neutral and selective diversity in plant species. Mathematical Modelling in Plant Biology, 195-212. https://doi.org/10.1007/978-3-319-99070-5_10 | Weak seed banks influence the signature and detectability of selective sweeps | Kevin Korfmann, Diala Abu Awad, Aurélien Tellier | <p style="text-align: justify;">Seed banking (or dormancy) is a widespread bet-hedging strategy, generating a form of population overlap, which decreases the magnitude of genetic drift. The methodological complexity of integrating this trait impli... | | Adaptation, Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Ecology, Genome Evolution, Life History, Population Genetics / Genomics | Renaud Vitalis | 2022-05-23 13:01:57 | ||
04 Mar 2024
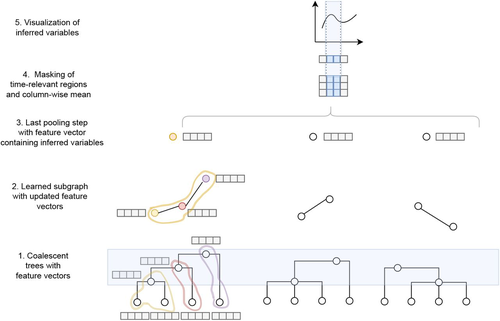
Simultaneous Inference of Past Demography and Selection from the Ancestral Recombination Graph under the Beta CoalescentBeyond the standard coalescent: demographic inference with complete genomes and graph neural networks under the beta coalescentRecommended by Julien Yann Dutheil based on reviews by 2 anonymous reviewers
Modelling the evolution of complete genome sequences in populations requires accounting for the recombination process, as a single tree can no longer describe the underlying genealogy. The sequentially Markov coalescent (SMC, McVean and Cardin 2005; Marjoram and Wall 2006) approximates the standard coalescent with recombination process and permits estimating population genetic parameters (e.g., population sizes, recombination rates) using population genomic datasets. As such datasets become available for an increasing number of species, more fine-tuned models are needed to encompass the diversity of life cycles of organisms beyond the model species on which most methods have been benchmarked. The work by Korfmann et al. (Korfmann et al. 2024) represents a significant step forward as it accounts for multiple mergers in SMC models. Multiple merger models account for simultaneous coalescence events so that more than two lineages find a common ancestor in a given generation. This feature is not allowed in standard coalescent models and may result from selection or skewed offspring distributions, conditions likely met by a broad range of species, particularly microbial. Yet, this work goes beyond extending the SMC, as it introduces several methodological innovations. The "classical" SMC-based inference approaches rely on hidden Markov models to compute the likelihood of the data while efficiently integrating over the possible ancestral recombination graphs (ARG). Following other recent works (e.g. Gattepaille et al. 2016), Korfmann et al. propose to separate the ARG inference from model parameter estimation under maximum likelihood (ML). They introduce a procedure where the ARG is first reconstructed from the data and then taken as input in the model fitting step. While this approach does not permit accounting for the uncertainty in the ARG reconstruction (which is typically large), it potentially allows for the extraction of more information from the ARG, such as the occurrence of multiple merging events. Going away from maximum likelihood inference, the authors trained a graph neural network (GNN) on simulated ARGs, introducing a new, flexible way to estimate population genomic parameters. The authors used simulations under a beta-coalescent model with diverse demographic scenarios and showed that the ML and GNN approaches introduced can reliably recover the simulated parameter values. They further show that when the true ARG is given as input, the GNN outperforms the ML approach, demonstrating its promising power as ARG reconstruction methods improve. In particular, they showed that trained GNNs can disentangle the effects of selective sweeps and skewed offspring distributions while inferring past population size changes. This work paves the way for new, exciting applications, though many questions must be answered. How frequent are multiple mergers? As the authors showed that these events "erase" the record of past demographic events, how many genomes are needed to conduct reliable inference, and can the methods computationally cope with the resulting (potentially large) amounts of required data? This is particularly intriguing as micro-organisms, prone to strong selection and skewed offspring distributions, also tend to carry smaller genomes. References Gattepaille L, Günther T, Jakobsson M. 2016. Inferring Past Effective Population Size from Distributions of Coalescent Times. Genetics 204:1191-1206. | Simultaneous Inference of Past Demography and Selection from the Ancestral Recombination Graph under the Beta Coalescent | Kevin Korfmann, Thibaut Sellinger, Fabian Freund, Matteo Fumagalli, Aurélien Tellier | <p style="text-align: justify;">The reproductive mechanism of a species is a key driver of genome evolution. The standard Wright-Fisher model for the reproduction of individuals in a population assumes that each individual produces a number of off... | | Adaptation, Bioinformatics & Computational Biology, Evolutionary Applications, Evolutionary Theory, Life History, Population Genetics / Genomics | Julien Yann Dutheil | 2023-07-31 13:11:22 | ||
19 Dec 2016

POSTPRINT
Geographic body size variation in the periodical cicadas Magicicada: implications for life cycle divergence and local adaptationMegacicadas show a temperature-mediated converse Bergmann cline in body size (larger in the warmer south) but no body size difference between 13- and 17-year species pairsRecommended by Wolf Blanckenhorn and Thomas FlattPeriodical cicadas are a very prominent insect group in North America that are known for their large size, good looks, and loud sounds. However, they are probably known best to evolutionary ecologists because of their long juvenile periods of 13 or 17 years (prime numbers!), which they spend in the ground. Multiple related species living in the same area are often coordinated in emerging as adults during the same year, thereby presumably swamping any predators specialized on eating them. Reference [1] Koyama T, Ito H, Kakishima S, Yoshimura J, Cooley JR, Simon C, Sota T. 2015. Geographic body size variation in the periodical cicadas Magicicada: implications for life cycle divergence and local adaptation. Journal of Evolutionary Biology 28:1270-1277. doi: 10.1111/jeb.12653 | Geographic body size variation in the periodical cicadas Magicicada: implications for life cycle divergence and local adaptation | Koyama T, Ito H, Kakishima S, Yoshimura J, Cooley JR, Simon C, Sota T | Seven species in three species groups (Decim, Cassini and Decula) of periodical cicadas (*Magicicada*) occupy a wide latitudinal range in the eastern United States. To clarify how adult body size, a key trait affecting fitness, varies geographical... | | Adaptation, Evolutionary Ecology, Life History, Macroevolution, Phylogeography & Biogeography, Speciation | Wolf Blanckenhorn | 2016-12-19 10:39:22 | ||
13 Dec 2016

POSTPRINT
A supergene determines highly divergent male reproductive morphs in the ruffSupergene Control of a Reproductive PolymorphismRecommended by Thomas Flatt and Laurent KellerTwo back-to-back papers published earlier this year in Nature Genetics provide compelling evidence for the control of a male reproductive polymorphism in a wading bird by a "supergene", a cluster of tightly linked genes [1-2]. The bird in question, the ruff (Philomachus pugnax), has a rather unusual reproductive system that consists of three distinct types of males ("reproductive morphs"): aggressive "independents" who represent the majority of males; a smaller fraction of non-territorial "satellites" who are submissive towards "independents"; and "faeders" who mimic females and are rare. Previous work has shown that the male morphs differ in major aspects of mating and aggression behavior, plumage coloration and body size, and that – intriguingly – this complex multi-trait polymorphism is apparently controlled by a single autosomal Mendelian locus with three alleles [3]. To uncover the genetic control of this polymorphism two independent teams, led by Terry Burke [1] and Leif Andersson [2], have set out to analyze the genomes of male ruffs. Using a combination of genomics and genetics, both groups managed to pin down the supergene locus and map it to a non-recombining, 4.5 Mb large inversion which arose 3.8 million years ago. While "independents" are homozygous for the ancestral uninverted sequence, "satellites" and "faeders" carry evolutionarily divergent, dominant alternative haplotypes of the inversion. Thus, as in several other notable cases, for example the supergene control of disassortative mating, aggressiveness and plumage color in white-throated sparrows [4], of mimicry in Heliconius and Papilio butterflies [5-6], or of social structure in ants [7], an inversion – behaving as a single "locus" – underpins the mechanistic basis of the supergene. More generally, and beyond inversions, a growing number of studies now shows that selection can favor the evolution of suppressed recombination, thereby leading to the emergence of clusters of tightly linked loci which can then control – presumably due to polygenic gene action – a suite of complex phenotypes [8-10]. A largely unresolved question in this field concerns the identity of the causative alleles and loci within a given supergene. Recent progress on this question has been made for example in Papilio polytes butterflies where a mimicry supergene has been found to involve – surprisingly – only a single but large gene: multiple mimicry alleles in the doublesex gene are maintained in strong linkage disequilibrium via an inversion. It will clearly be of great interest to see future examples of such a fine-scale genetic dissection of supergenes. In conclusion, we were impressed by the data and analyses of Küpper et al. [1] and Lamichhaney et al. [2]: both papers beautifully illustrate how genomics and evolutionary ecology can be combined to make new, exciting discoveries. Both papers will appeal to readers with an interest in supergenes, inversions, the interplay of selection and recombination, or the genetic control of complex phenotypes. References [1] Küpper C, Stocks M, Risse JE, dos Remedios N, Farrell LL, McRae SB, Morgan TC, Karlionova N, Pinchuk P, Verkuil YI, et al. 2016. A supergene determines highly divergent male reproductive morphs in the ruff. Nature Genetics 48:79-83. doi: 10.1038/ng.3443 [2] Lamichhaney S, Fan G, Widemo F, Gunnarsson U, Thalmann DS, Hoeppner MP, Kerje S, Gustafson U, Shi C, Zhang H, et al. 2016. Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax). Nature Genetics 48:84-88. doi: 10.1038/ng.3430 [3] Lank DB, Smith CM, Hanotte O, Burke T, Cooke F. 1995. Genetic polymorphism for alternative mating behaviour in lekking male ruff Philomachus pugnax. Nature 378:59-62. doi: 10.1038/378059a0 [4] Tuttle Elaina M, Bergland Alan O, Korody Marisa L, Brewer Michael S, Newhouse Daniel J, Minx P, Stager M, Betuel A, Cheviron Zachary A, Warren Wesley C, et al. 2016. Divergence and Functional Degradation of a Sex Chromosome-like Supergene. Current Biology 26:344-350. doi: 10.1016/j.cub.2015.11.069 [5] Joron M, Frezal L, Jones RT, Chamberlain NL, Lee SF, Haag CR, Whibley A, Becuwe M, Baxter SW, Ferguson L, et al. 2011. Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477:203-206. doi: 10.1038/nature10341 [6] Kunte K, Zhang W, Tenger-Trolander A, Palmer DH, Martin A, Reed RD, Mullen SP, Kronforst MR. 2014. doublesex is a mimicry supergene. Nature 507:229-232. doi: 10.1038/nature13112 [7] Wang J, Wurm Y, Nipitwattanaphon M, Riba-Grognuz O, Huang Y-C, Shoemaker D, Keller L. 2013. A Y-like social chromosome causes alternative colony organization in fire ants. Nature 493:664-668. doi: 10.1038/nature11832 [8] Thompson MJ, Jiggins CD. 2014. Supergenes and their role in evolution. Heredity 113:1-8. doi: 10.1038/hdy.2014.20 [9] Schwander T, Libbrecht R, Keller L. 2014. Supergenes and Complex Phenotypes. Current Biology 24:R288-R294. doi: 10.1016/j.cub.2014.01.056 [10] Charlesworth D. 2015. The status of supergenes in the 21st century: recombination suppression in Batesian mimicry and sex chromosomes and other complex adaptations. Evolutionary Applications 9:74-90. doi: 10.1111/eva.12291 | A supergene determines highly divergent male reproductive morphs in the ruff | Küpper C, Stocks M, Risse JE, dos Remedios N, Farrell LL, McRae SB, Morgan TC, Karlionova N, Pinchuk P, Verkuil YI, et al. | Three strikingly different alternative male mating morphs (aggressive 'independents', semicooperative 'satellites' and female-mimic 'faeders') coexist as a balanced polymorphism in the ruff, *Philomachus pugnax*, a lek-breeding wading bird1, 2, 3.... | | Adaptation, Genotype-Phenotype, Life History, Population Genetics / Genomics, Reproduction and Sex | Thomas Flatt | 2016-12-13 17:28:13 | ||
13 Dec 2016

POSTPRINT
Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax)Supergene Control of a Reproductive PolymorphismRecommended by Thomas Flatt and Laurent KellerTwo back-to-back papers published earlier this year in Nature Genetics provide compelling evidence for the control of a male reproductive polymorphism in a wading bird by a "supergene", a cluster of tightly linked genes [1-2]. The bird in question, the ruff (Philomachus pugnax), has a rather unusual reproductive system that consists of three distinct types of males ("reproductive morphs"): aggressive "independents" who represent the majority of males; a smaller fraction of non-territorial "satellites" who are submissive towards "independents"; and "faeders" who mimic females and are rare. Previous work has shown that the male morphs differ in major aspects of mating and aggression behavior, plumage coloration and body size, and that – intriguingly – this complex multi-trait polymorphism is apparently controlled by a single autosomal Mendelian locus with three alleles [3]. To uncover the genetic control of this polymorphism two independent teams, led by Terry Burke [1] and Leif Andersson [2], have set out to analyze the genomes of male ruffs. Using a combination of genomics and genetics, both groups managed to pin down the supergene locus and map it to a non-recombining, 4.5 Mb large inversion which arose 3.8 million years ago. While "independents" are homozygous for the ancestral uninverted sequence, "satellites" and "faeders" carry evolutionarily divergent, dominant alternative haplotypes of the inversion. Thus, as in several other notable cases, for example the supergene control of disassortative mating, aggressiveness and plumage color in white-throated sparrows [4], of mimicry in Heliconius and Papilio butterflies [5-6], or of social structure in ants [7], an inversion – behaving as a single "locus" – underpins the mechanistic basis of the supergene. More generally, and beyond inversions, a growing number of studies now shows that selection can favor the evolution of suppressed recombination, thereby leading to the emergence of clusters of tightly linked loci which can then control – presumably due to polygenic gene action – a suite of complex phenotypes [8-10]. A largely unresolved question in this field concerns the identity of the causative alleles and loci within a given supergene. Recent progress on this question has been made for example in Papilio polytes butterflies where a mimicry supergene has been found to involve – surprisingly – only a single but large gene: multiple mimicry alleles in the doublesex gene are maintained in strong linkage disequilibrium via an inversion. It will clearly be of great interest to see future examples of such a fine-scale genetic dissection of supergenes. In conclusion, we were impressed by the data and analyses of Küpper et al. [1] and Lamichhaney et al. [2]: both papers beautifully illustrate how genomics and evolutionary ecology can be combined to make new, exciting discoveries. Both papers will appeal to readers with an interest in supergenes, inversions, the interplay of selection and recombination, or the genetic control of complex phenotypes. References [1] Küpper C, Stocks M, Risse JE, dos Remedios N, Farrell LL, McRae SB, Morgan TC, Karlionova N, Pinchuk P, Verkuil YI, et al. 2016. A supergene determines highly divergent male reproductive morphs in the ruff. Nature Genetics 48:79-83. doi: 10.1038/ng.3443 [2] Lamichhaney S, Fan G, Widemo F, Gunnarsson U, Thalmann DS, Hoeppner MP, Kerje S, Gustafson U, Shi C, Zhang H, et al. 2016. Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax). Nature Genetics 48:84-88. doi: 10.1038/ng.3430 [3] Lank DB, Smith CM, Hanotte O, Burke T, Cooke F. 1995. Genetic polymorphism for alternative mating behaviour in lekking male ruff Philomachus pugnax. Nature 378:59-62. doi: 10.1038/378059a0 [4] Tuttle Elaina M, Bergland Alan O, Korody Marisa L, Brewer Michael S, Newhouse Daniel J, Minx P, Stager M, Betuel A, Cheviron Zachary A, Warren Wesley C, et al. 2016. Divergence and Functional Degradation of a Sex Chromosome-like Supergene. Current Biology 26:344-350. doi: 10.1016/j.cub.2015.11.069 [5] Joron M, Frezal L, Jones RT, Chamberlain NL, Lee SF, Haag CR, Whibley A, Becuwe M, Baxter SW, Ferguson L, et al. 2011. Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477:203-206. doi: 10.1038/nature10341 [6] Kunte K, Zhang W, Tenger-Trolander A, Palmer DH, Martin A, Reed RD, Mullen SP, Kronforst MR. 2014. doublesex is a mimicry supergene. Nature 507:229-232. doi: 10.1038/nature13112 [7] Wang J, Wurm Y, Nipitwattanaphon M, Riba-Grognuz O, Huang Y-C, Shoemaker D, Keller L. 2013. A Y-like social chromosome causes alternative colony organization in fire ants. Nature 493:664-668. doi: 10.1038/nature11832 [8] Thompson MJ, Jiggins CD. 2014. Supergenes and their role in evolution. Heredity 113:1-8. doi: 10.1038/hdy.2014.20 [9] Schwander T, Libbrecht R, Keller L. 2014. Supergenes and Complex Phenotypes. Current Biology 24:R288-R294. doi: 10.1016/j.cub.2014.01.056 [10] Charlesworth D. 2015. The status of supergenes in the 21st century: recombination suppression in Batesian mimicry and sex chromosomes and other complex adaptations. Evolutionary Applications 9:74-90. doi: 10.1111/eva.12291 | Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax) | Lamichhaney S, Fan G, Widemo F, Gunnarsson U, Thalmann DS, Hoeppner MP, Kerje S, Gustafson U, Shi C, Zhang H, et al. | The ruff is a Palearctic wader with a spectacular lekking behavior where highly ornamented males compete for females1, 2, 3, 4. This bird has one of the most remarkable mating systems in the animal kingdom, comprising three different male morphs (... | | Adaptation, Behavior & Social Evolution, Genotype-Phenotype, Life History, Population Genetics / Genomics, Quantitative Genetics, Reproduction and Sex | Thomas Flatt | 2016-12-13 17:46:54 |




