
Latest recommendations

| Id▲ | Title * | Authors * | Abstract * | Picture * | Thematic fields * | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
20 Sep 2017

An interaction between cancer progression and social environment in DrosophilaCancer and loneliness in DrosophilaRecommended by Ana Rivero based on reviews by Ana Rivero and Silvie HuijbenDrosophila flies may not be perceived as a quintessentially social animal, particularly when compared to their eusocial hymenopteran cousins. Although they have no parental care, division of labour or subfertile caste, fruit flies nevertheless exhibit an array of social phenotypes that are potentially comparable to those of their highly social relatives. In the wild, Drosophila adults cluster around food resources where courtship, mating activity and oviposition occur. Recent work has shown not only that social interactions in these clusters condition many aspects of the behaviour and physiology of the flies [1] but also, and perhaps more unexpectedly, that social isolation has a negative impact on their fitness [2]. Many studies in humans point to the role of social isolation as a source of stress that can induce and accelerate disease progression. The ultimate proof of the connection between social interaction and disease is however mired in confounding variables and alternative explanations so the subject, though crucial, remains controversial. With a series of elegant experiments using Drosophila flies that develop an inducible form of intestinal cancer, Dawson et al [3] show that cancer progresses more rapidly in flies maintained in isolation than in flies maintained with other cancerous flies. Further, cancerous flies kept with non-cancerous flies, fare just as badly as when kept alone. Their experiments suggest that this is due to the combined effect of healthy flies avoiding contact with cancerous flies (even though this is a non-contagious disease), and of cancerous flies having higher quality interactions with other cancerous flies than with healthy ones. Perceived isolation is therefore as pernicious as real isolation when it comes to cancer progression in these flies. Like all good research, this study opens up as many questions as it answers, in particular the why and wherefores of the flies’ extraordinary social behaviour in the face of disease. References [1] Camiletti AL and Thompson GJ. 2016. Drosophila as a genetically tractable model for social insect behavior. Frontiers in Ecology and Evolution, 4: 40. doi: 10.3389/fevo.2016.00040 [2] Ruan H and Wu C-F. 2008. Social interaction-mediated lifespan extension of Drosophila Cu/Zn superoxide dismutase mutants. Proceedings of the National Academy of Sciences, USA, 105: 7506-7510. doi: 10.1073/pnas.0711127105 [3] Dawson E, Bailly T, Dos Santos J, Moreno C, Devilliers M, Maroni B, Sueur C, Casali A, Ujvari B, Thomas F, Montagne J, Mery F. 2017. An interaction between cancer progression and social environment in Drosophila. BiorXiv, 143560, ver. 3 of 19th September 2017. doi: 10.1101/143560 | An interaction between cancer progression and social environment in Drosophila | Erika H. Dawson, Tiphaine P.M. Bailly, Julie Dos Santos , Céline Moreno, Maëlle Devilliers, Brigitte Maroni, Cédric Sueur, Andreu Casali, Beata Ujvari, Frederic Thomas, Jacques Montagne, Frederic Mery | The ecological benefits of sociality in gregarious species are widely acknowledged. However, only limited data is available on how the social environment influences non-communicable disease outcomes. For instance, despite extensive research over t... | | Behavior & Social Evolution, Evolutionary Ecology, Phenotypic Plasticity | Ana Rivero | 2017-05-30 08:55:16 | ||
29 Sep 2017

Parallel diversifications of Cremastosperma and Mosannona (Annonaceae), tropical rainforest trees tracking Neogene upheaval of the South American continentUnravelling the history of Neotropical plant diversificationRecommended by Hervé Sauquet based on reviews by Thomas Couvreur and Hervé SauquetSouth American rainforests, particularly the Tropical Andes, have been recognized as the hottest spot of plant biodiversity on Earth, while facing unprecedented threats from human impact [1,2]. Considerable research efforts have recently focused on unravelling the complex geological, bioclimatic, and biogeographic history of the region [3,4]. While many studies have addressed the question of Neotropical plant diversification using parametric methods to reconstruct ancestral areas and patterns of dispersal, Pirie et al. [5] take a distinct, complementary approach. Based on a new, near-complete molecular phylogeny of two Neotropical genera of the flowering plant family Annonaceae, the authors modelled the ecological niche of each species and reconstructed the history of niche differentiation across the region. The main conclusion is that, despite similar current distributions and close phylogenetic distance, the two genera experienced rather distinct processes of diversification, responding differently to the major geological events marking the history of the region in the last 20 million years (Andean uplift, drainage of Lake Pebas, and closure of the Panama Isthmus). As a researcher who has not personally worked on Neotropical biogeography, I found this paper captivating and especially enjoyed very much reading the Introduction, which sets out the questions very clearly. The strength of this paper is the near-complete diversity of species the authors were able to sample in each clade and the high-quality data compiled for the niche models. I would recommend this paper as a nice example of a phylogenetic study aimed at unravelling the detailed history of Neotropical plant diversification. While large, synthetic meta-analyses of many clades should continue to seek general patterns [4,6], careful studies restricted on smaller, but well controlled and sampled datasets such as this one are essential to really understand tropical plant diversification in all its complexity. References [1] Antonelli A, and Sanmartín I. 2011. Why are there so many plant species in the Neotropics? Taxon 60, 403–414. [2] Mittermeier RA, Robles-Gil P, Hoffmann M, Pilgrim JD, Brooks TB, Mittermeier CG, Lamoreux JL and Fonseca GAB. 2004. Hotspots revisited: Earths biologically richest and most endangered ecoregions. CEMEX, Mexico City, Mexico 390pp [3] Antonelli A, Nylander JAA, Persson C and Sanmartín I. 2009. Tracing the impact of the Andean uplift on Neotropical plant evolution. Proceedings of the National Academy of Science of the USA 106, 9749–9754. doi: 10.1073/pnas.0811421106 [4] Hoorn C, Wesselingh FP, ter Steege H, Bermudez MA, Mora A, Sevink J, Sanmartín I, Sanchez-Meseguer A, Anderson CL, Figueiredo JP, Jaramillo C, Riff D, Negri FR, Hooghiemstra H, Lundberg J, Stadler T, Särkinen T and Antonelli A. 2010. Amazonia through time: Andean uplift, climate change, landscape evolution, and biodiversity. Science 330, 927–931. doi: 10.1126/science.1194585 [5] Pirie MD, Maas PJM, Wilschut R, Melchers-Sharrott H and Chatrou L. 2017. Parallel diversifications of Cremastosperma and Mosannona (Annonaceae), tropical rainforest trees tracking Neogene upheaval of the South American continent. bioRxiv, 141127, ver. 3 of 28th Sept 2017. doi: 10.1101/141127 [6] Bacon CD, Silvestro D, Jaramillo C, Tilston Smith B, Chakrabartye P and Antonelli A. 2015. Biological evidence supports an early and complex emergence of the Isthmus of Panama. Proceedings of the National Academy of Science of the USA 112, 6110–6115. doi: 10.1073/pnas.1423853112 | Parallel diversifications of Cremastosperma and Mosannona (Annonaceae), tropical rainforest trees tracking Neogene upheaval of the South American continent | Michael D. Pirie, Paul J. M. Maas, Rutger A. Wilschut, Heleen Melchers-Sharrott & Lars W. Chatrou | Much of the immense present day biological diversity of Neotropical rainforests originated from the Miocene onwards, a period of geological and ecological upheaval in South America. We assess the impact of the Andean orogeny, drainage of lake Peba... | | Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Hervé Sauquet | Hervé Sauquet, Thomas Couvreur | 2017-06-03 21:25:48 | |
05 Oct 2017

Using Connectivity To Identify Climatic Drivers Of Local AdaptationA new approach to identifying drivers of local adaptationRecommended by Ruth Arabelle Hufbauer based on reviews by Ruth Arabelle Hufbauer and Thomas LenormandLocal adaptation, the higher fitness a population achieves in its local “home” environment relative to other environments is a crucial phase in the divergence of populations, and as such both generates and maintains diversity. Local adaptation is enhanced by selection and genetic variation in the relevant traits, and decreased by gene flow and genetic drift. Demonstrating local adaptation is laborious, and is typically done with a reciprocal transplant design [1], documenting repeated geographic clines [e.g. 2, 3] also provides strong evidence of local adaptation. Even when well documented, it is often unknown which aspects of the environment impose selection. Indeed, differences in environment between different sites that are measured during studies of local adaptation explain little of the variance in the degree of local adaptation [4]. This poses a problem to population management. Given climate change and habitat destruction, understanding the environmental drivers of local adaptation can be crucially important to conducting successful assisted migration or targeted gene flow. In this manuscript, Macdonald et al. [5] propose a means of identifying which aspects of the environment select for local adaptation without conducting a reciprocal transplant experiment. The idea is that the strength of relationships between traits and environmental variables that are due to plastic responses to the environment will not be influenced by gene flow, but the strength of trait-environment relationships that are due to local adaptation should decrease with gene flow. This then can be used to reduce the somewhat arbitrary list of environmental variables on which data are available down to a targeted list more likely to drive local adaptation in specific traits. To perform such an analysis requires three things: 1) measurements of traits of interest in a species across locations, 2) an estimate of gene flow between locations, which can be replaced with a biologically meaningful estimate of how well connected those locations are from the point of view of the study species, and 3) data on climate and other environmental variables from across a species’ range, many of which are available on line. Macdonald et al. [5] demonstrate their approach using a skink (Lampropholis coggeri). They collected morphological and physiological data on individuals from multiple populations. They estimated connectivity among those locations using information on habitat suitability and dispersal potential [6], and gleaned climatic data from available databases and the literature. They find that two physiological traits, the critical minimum and maximum temperatures, show the strongest signs of local adaptation, specifically local adaptation to annual mean precipitation, precipitation of the driest quarter, and minimum annual temperature. These are then aspects of skink phenotype and skink habitats that could be explored further, or could be used to provide background information if migration efforts, for example for genetic rescue [7] were initiated. The approach laid out has the potential to spark a novel genre of research on local adaptation. It its simplest form, knowing that local adaptation is eroded by gene flow, it is intuitive to consider that if connectivity reduces the strength of the relationship between an environmental variable and a trait, that the trait might be involved in local adaptation. The approach is less intuitive than that, however – it relies not connectivity per-se, but the interaction between connectivity and different environmental variables and how that interaction alters trait-environment relationships. The authors lay out a number of useful caveats and potential areas that could use further development. It will be interesting to see how the community of evolutionary biologists responds. References [1] Blanquart F, Kaltz O, Nuismer SL and Gandon S. 2013. A practical guide to measuring local adaptation. Ecology Letters, 16: 1195-1205. doi: 10.1111/ele.12150 [2] Huey RB, Gilchrist GW, Carlson ML, Berrigan D and Serra L. 2000. Rapid evolution of a geographic cline in size in an introduced fly. Science, 287: 308-309. doi: 10.1126/science.287.5451.308 [3] Milesi P, Lenormand T, Lagneau C, Weill M and Labbé P. 2016. Relating fitness to long-term environmental variations in natura. Molecular Ecology, 25: 5483-5499. doi: 10.1111/mec.13855 [4] Hereford, J. 2009. A quantitative survey of local adaptation and fitness trade-offs. The American Naturalist 173: 579-588. doi: 10.1086/597611 [5] Macdonald SL, Llewelyn J and Phillips BL. 2017. Using connectivity to identify climatic drivers of local adaptation. bioRxiv, ver. 4 of October 4, 2017. doi: 10.1101/145169 [6] Macdonald SL, Llewelyn J, Moritz C and Phillips BL. 2017. Peripheral isolates as sources of adaptive diversity under climate change. Frontiers in Ecology and Evolution, 5:88. doi: 10.3389/fevo.2017.00088 [7] Whiteley AR, Fitzpatrick SW, Funk WC and Tallmon DA. 2015. Genetic rescue to the rescue. Trends in Ecology & Evolution, 30: 42-49. doi: 10.1016/j.tree.2014.10.009 | Using Connectivity To Identify Climatic Drivers Of Local Adaptation | Stewart L. Macdonald, John Llewelyn, Ben Phillips | Despite being able to conclusively demonstrate local adaptation, we are still often unable to objectively determine the climatic drivers of local adaptation. Given the rapid rate of global change, understanding the climatic drivers of local adapta... | | Adaptation, Evolutionary Applications | Ruth Arabelle Hufbauer | Thomas Lenormand | 2017-06-06 13:06:54 | |
20 Dec 2017

Renewed diversification following Miocene landscape turnover in a Neotropical butterfly radiationThe influence of environmental change over geological time on the tempo and mode of biological diversification, revealed by Neotropical butterfliesRecommended by Richard H Ree based on reviews by Delano Lewis and 1 anonymous reviewerThe influence of environmental change over geological time on the tempo and mode of biological diversification is a hot topic in biogeography. Of central interest are questions about where, when, and how fast lineages proliferated, suffered extinction, and migrated in response to tectonic events, the waxing and waning of dominant biomes, etc. In this context, the dynamic conditions of the Miocene have received much attention, from studies of many clades and biogeographic regions. Here, Chazot et al. [1] present an exemplary analysis of butterflies (tribe Ithomiini) in the Neotropics, examining their diversification across the Andes and Amazon. They infer sharp contrasts between these regions in the late Miocene: accelerated diversification during orogeny of the Andes, and greater extinction in the Amazon associated during the Pebas system, with interchange and local diversification increasing following the Pebas during the Pliocene. References [1] Chazot N, Willmott KR, Lamas G, Freitas AVL, Piron-Prunier F, Arias CF, Mallet J, De-Silva DL and Elias M. 2017. Renewed diversification following Miocene landscape turnover in a Neotropical butterfly radiation. BioRxiv 148189, ver 4 of 19th December 2017. doi: 10.1101/148189 [2] Xing Y, and Ree RH. 2017. Uplift-driven diversification in the Hengduan Mountains, a temperate biodiversity hotspot. Proceedings of the National Academy of Sciences of the United States of America, 114: E3444-E3451. doi: 10.1073/pnas.1616063114 | Renewed diversification following Miocene landscape turnover in a Neotropical butterfly radiation | Nicolas Chazot, Keith R. Willmott, Gerardo Lamas, André V.L. Freitas, Florence Piron-Prunier, Carlos F. Arias, James Mallet, Donna Lisa De-Silva, Marianne Elias | The Neotropical region has experienced a dynamic landscape evolution throughout the Miocene, with the large wetland Pebas occupying western Amazonia until 11-8 my ago and continuous uplift of the Andes mountains along the western edge of South Ame... | | Macroevolution, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Richard H Ree | 2017-06-12 11:55:14 | ||
18 Dec 2017
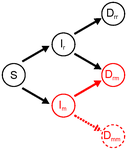
Co-evolution of virulence and immunosuppression in multiple infectionsTwo parasites, virulence and immunosuppression: how does the whole thing evolve?Recommended by Sara Magalhaes based on reviews by 2 anonymous reviewersHow parasite virulence evolves is arguably the most important question in both the applied and fundamental study of host-parasite interactions. Typically, this research area has been progressing through the formalization of the problem via mathematical modelling. This is because the question is a complex one, as virulence is both affected and affects several aspects of the host-parasite interaction. Moreover, the evolution of virulence is a problem in which ecology (epidemiology) and evolution (changes in trait values through time) are tightly intertwined, generating what is now known as eco-evolutionary dynamics. Therefore, intuition is not sufficient to address how virulence may evolve. References [1] Anderson RM and May RM. 1982. Coevolution of hosts and parasites. Parasitology, 1982. 85: 411–426. doi: 10.1017/S0031182000055360 [2] Kamiya T, Mideo N and Alizon S. 2017. Coevolution of virulence and immunosuppression in multiple infections. bioRxiv, ver. 7 peer-reviewed by PCI Evol Biol, 149211. doi: 10.1101/139147 | Co-evolution of virulence and immunosuppression in multiple infections | Tsukushi Kamiya, Nicole Mideo, Samuel Alizon | Many components of the host-parasite interaction have been shown to affect the way virulence, that is parasite induced harm to the host, evolves. However, co-evolution of multiple traits is often neglected. We explore how an immunosuppressive mech... | | Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory | Sara Magalhaes | 2017-06-13 16:49:45 | ||
05 Jun 2018

Pleistocene climate change and the formation of regional species poolsRecent assembly of European biogeographic species poolRecommended by Fabien Condamine based on reviews by 3 anonymous reviewersBiodiversity is unevenly distributed over time, space and the tree of life [1]. The fact that regions are richer than others as exemplified by the latitudinal diversity gradient has fascinated biologists as early as the first explorers travelled around the world [2]. Provincialism was one of the first general features of land biotic distributions noted by famous nineteenth century biologists like the phytogeographers J.D. Hooker and A. de Candolle, and the zoogeographers P.L. Sclater and A.R. Wallace [3]. When these explorers travelled among different places, they were struck by the differences in their biotas (e.g. [4]). The limited distributions of distinctive endemic forms suggested a history of local origin and constrained dispersal. Much biogeographic research has been devoted to identifying areas where groups of organisms originated and began their initial diversification [3]. Complementary efforts found evidence of both historical barriers that blocked the exchange of organisms between adjacent regions and historical corridors that allowed dispersal between currently isolated regions. The result has been a division of the Earth into a hierarchy of regions reflecting patterns of faunal and floral similarities (e.g. regions, subregions, provinces). Therefore a first ensuing question is: “how regional species pools have been assembled through time and space?”, which can be followed by a second question: “what are the ecological and evolutionary processes leading to differences in species richness among species pools?”. To address these questions, the study of Calatayud et al. [5] developed and performed an interesting approach relying on phylogenetic data to identify regional and sub-regional pools of European beetles (using the iconic ground beetle genus Carabus). Specifically, they analysed the processes responsible for the assembly of species pools, by comparing the effects of dispersal barriers, niche similarities and phylogenetic history. They found that Europe could be divided in seven modules that group zoogeographically distinct regions with their associated faunas, and identified a transition zone matching the limit of the ice sheets at Last Glacial Maximum (19k years ago). Deviance of species co-occurrences across regions, across sub-regions and within each region was significantly explained, primarily by environmental niche similarity, and secondarily by spatial connectivity, except for northern regions. Interestingly, southern species pools are mostly separated by dispersal barriers, whereas northern species pools are mainly sorted by their environmental niches. Another important finding of Calatayud et al. [5] is that most phylogenetic structuration occurred during the Pleistocene, and they show how extreme recent historical events (Quaternary glaciations) can profoundly modify the composition and structure of geographic species pools, as opposed to studies showing the role of deep-time evolutionary processes. The study of biogeographic assembly of species pools using phylogenies has never been more exciting and promising than today. Catalayud et al. [5] brings a nice study on the importance of Pleistocene glaciations along with geographical barriers and niche-based processes in structuring the regional faunas of European beetles. The successful development of powerful analytical tools in recent years, in conjunction with the rapid and massive increase in the availability of biological data (including molecular phylogenies, fossils, georeferrenced occurrences and ecological traits), will allow us to disentangle complex evolutionary histories. Although we still face important limitations in data availability and methodological shortcomings, the last decade has witnessed an improvement of our understanding of how historical and biotic triggers are intertwined on shaping the Earth’s stupendous biological diversity. I hope that the Catalayud et al.’s approach (and analytical framework) will help movement in that direction, and that it will provide interesting perspectives for future investigations of other regions. Applied to a European beetle radiation, they were able to tease apart the relative contributions of biotic (niche-based processes) versus abiotic (geographic barriers and climate change) factors. References [1] Rosenzweig ML. 1995. Species diversity in space and time. Cambridge: Cambridge University Press. | Pleistocene climate change and the formation of regional species pools | Joaquín Calatayud, Miguel Á. Rodríguez, Rafael Molina-Venegas, María Leo, José Luís Hórreo, Joaquín Hortal | <p>Despite the description of bioregions dates back from the origin of biogeography, the processes originating their associated species pools have been seldom studied. Ancient historical events are thought to play a fundamental role in configuring... | | Phylogeography & Biogeography | Fabien Condamine | 2017-06-14 07:30:32 | ||
12 Jul 2017

Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategiesTowards an integrated scenario to understand evolutionary patterns in A. thalianaRecommended by Xavier Picó based on reviews by Rafa Rubio de Casas and Xavier PicóNobody can ignore that a full understanding of evolution requires an integrated approach from both conceptual and methodological viewpoints. Although some life-history traits, e.g. flowering time, have long been receiving more attention than others, in many cases because the former are more workable than the latter, we must acknowledge that our comprehension about how evolution works is strongly biased and limited. In the Arabidopsis community, such an integration is making good progress as an increasing number of research groups worldwide are changing the way in which evolution is put to the test. This manuscript [1] is a good example of that as the authors raise an important issue in evolutionary biology by combining gene expression and flowering time data from different sources. In particular, the authors explore how variation in flowering time, which determines lifespan, and host immunity defenses co-vary, which is interpreted in terms of co-evolution between the two traits. Interestingly, the authors go beyond that pattern by separating lifespan-dependent from lifespan–independent defense genes, and by showing that defense genes with variants known to impact fitness in the field are among the genes whose expression co-varies most strongly with flowering time. Finally, these results are supported by a simple mathematical model indicating that such a relationship can also be expected theoretically. Overall, the readers will find many conceptual and methodological elements of interest in this manuscript. The idea that evolution is better understood under the scope of life history variation is really exciting and challenging, and in my opinion on the right track for disentangling the inherent complexities of evolutionary research. However, only when we face complexity, we also face its costs and burdens. In this particular case, the well-known co-variation between seed dormancy and flowering time is a missing piece, as well as the identification of (variation in) putative selective pressures accounting for the co-evolution between defense mechanisms and life history (seed dormancy vs. flowering time) along environmental gradients. More intellectual, technical and methodological challenges that with no doubt are totally worth it. Reference [1] Glander S, He F, Schmitz G, Witten A, Telschow A, de Meaux J. 2017. Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategies. bioRxiv ver.1 of June 19, 2017. doi: 10.1101/131136 | Assortment of flowering time and defense alleles in natural Arabidopsis thaliana populations suggests co-evolution between defense and vegetative lifespan strategies | Glander S, He F, Schmitz G, Witten A, Telschow A, de Meaux J | The selective impact of pathogen epidemics on host defenses can be strong but remains transient. By contrast, life-history shifts can durably and continuously modify the balance between costs and benefits of immunity, which arbitrates the evolutio... | | Adaptation, Evolutionary Ecology, Expression Studies, Life History, Phenotypic Plasticity, Quantitative Genetics, Species interactions | Xavier Picó | Sophie Karrenberg, Rafa Rubio de Casas, Xavier Picó | 2017-06-21 10:57:14 | |
13 Nov 2017

Epidemiological trade-off between intra- and interannual scales in the evolution of aggressiveness in a local plant pathogen populationThe pace of pathogens’ adaptation to their host plantsRecommended by Benoit Moury based on reviews by Benoit Moury and 1 anonymous reviewerBecause of their shorter generation times and larger census population sizes, pathogens are usually ahead in the evolutionary race with their hosts. The risks linked to pathogen adaptation are still exacerbated in agronomy, where plant and animal populations are not freely evolving but depend on breeders and growers, and are usually highly genetically homogeneous. As a consequence, the speed of pathogen adaptation is crucial for agriculture sustainability. Unraveling the time scale required for pathogens’ adaptation to their hosts would notably greatly improve our estimation of the risks of pathogen emergence, the efficiency of disease control strategies and the design of epidemiological surveillance schemes. However, the temporal scale of pathogen evolution has received much less attention than its spatial scale [1]. In their study of a wheat fungal disease, Suffert et al. [2] reached contrasting conclusions about the pathogen adaptation depending on the time scale (intra- or inter-annual) and on the host genotype (sympatric or allopatric) considered, questioning the experimental assessment of this important problem. Suffert et al. [2] sampled two pairs of Zymoseptoria tritici (the causal agent of septoria leaf blotch) sub-populations in a bread wheat field plot, representing (i) isolates collected at the beginning or at the end of an epidemic in a single growing season (2009-2010 intra-annual sampling scale) and (ii) isolates collected from plant debris at the end of growing seasons in 2009 and in 2015 (inter-annual sampling scale). Then, they measured in controlled conditions two aggressiveness traits of the isolates of these four Z. tritici sub-populations, the latent period and the lesion size on leaves, on two wheat cultivars. One of the cultivars was considered as "sympatric" because it was at the source of the studied isolates and was predominant in the growing area before the experiment, whereas the other cultivar was considered as "allopatric" since it replaced the previous one and became predominant in the growing area during the sampling period. On the sympatric host, at the intra-annual scale, they observed a marginally-significant decrease in latent period and a significant decrease of the between-isolate variance for this trait, which are consistent with a selection of pathogen variants with an enhanced aggressiveness. In contrast, at the inter-annual scale, no difference in the mean or variance of aggressiveness trait values was observed on the sympatric host, suggesting a lack of pathogen adaptation. They interpreted the contrast between observations at the two time scales as the consequence of a trade-off for the pathogen between a gain of aggressiveness after several generations of asexual reproduction at the intra-annual scale and a decrease of the probability to reproduce sexually and to be transmitted from one growing season to the next. Indeed, at the end of the growing season, the most aggressive isolates are located on the upper leaves of plants, where the pathogen density and hence probably also the probability to reproduce sexually, is lower. On the allopatric host, the conclusion about the pathogen stability at the inter-annual scale was somewhat different, since a significant increase in the mean lesion size was observed (isolates corresponding to the intra-annual scale were not checked on the allopatric host). This shows the possibility for the pathogen to evolve at the inter-annual scale, for a given aggressiveness trait and on a given host. In conclusion, Suffert et al.’s [2] study emphasizes the importance of the experimental design in terms of sampling time scale and host genotype choice to analyze the pathogen adaptation to its host plants. It provides also an interesting scenario, at the crossroad of the pathogen’s reproduction regime, niche partitioning and epidemiological processes, to interpret these contrasted results. Pathogen adaptation to plant cultivars with major-effect resistance genes is usually fast, including in the wheat-Z. tritici system [3]. Therefore, this study will be of great help for future studies on pathogen adaptation to plant partial resistance genes and on strategies of deployment of such resistance at the landscape scale. References [2] Suffert F, Goyeau H, Sache I, Carpentier F, Gelisse S, Morais D and Delestre G. 2017. Epidemiological trade-off between intra- and interannual scales in the evolution of aggressiveness in a local plant pathogen population. bioRxiv, 151068, ver. 3 of 12th November 2017. doi: 10.1101/151068 [3] Brown JKM, Chartrain L, Lasserre-Zuber P and Saintenac C. 2015. Genetics of resistance to Zymoseptoria tritici and applications to wheat breeding. Fungal Genetics and Biology, 79: 33–41. doi: 10.1016/j.fgb.2015.04.017 | Epidemiological trade-off between intra- and interannual scales in the evolution of aggressiveness in a local plant pathogen population | Frederic Suffert, Henriette Goyeau, Ivan Sache, Florence Carpentier, Sandrine Gelisse, David Morais, Ghislain Delestre | The efficiency of plant resistance to fungal pathogen populations is expected to decrease over time, due to its evolution with an increase in the frequency of virulent or highly aggressive strains. This dynamics may differ depending on the scale i... | | Adaptation, Evolutionary Applications, Evolutionary Epidemiology | Benoit Moury | 2017-06-23 21:04:54 | ||
07 Nov 2017

MaxTiC: Fast ranking of a phylogenetic tree by Maximum Time Consistency with lateral gene transfersDating nodes in a phylogeny using inferred horizontal gene transfersRecommended by Tatiana Giraud and Toni Gabaldon based on reviews by Alexandros Stamatakis, Mukul Bansal and 2 anonymous reviewersDating nodes in a phylogeny is an important problem in evolution and is typically performed by using molecular clocks and fossil age estimates [1]. The manuscript by Chauve et al. [2] reports a novel method, which uses lateral gene transfers to help ordering nodes in a species tree. The idea is that a lateral gene transfer can only occur between two species living at the same time, which indirectly informs on node relative ages in a phylogeny: the donor species cannot be more recent than the recipient species. Horizontal gene transfers are increasingly recognized as frequent, even in eukaryotes, and especially in micro-organisms that have little fossil records [3-7]. Yet, such an important source of information has been very rarely used so far for inferring relative node ages in phylogenies. In this context, the method by Chauve et al. [2] represents an innovative and original approach to a difficult problem. An obvious limitation of the approach is that it relies on inferences of horizontal transfers, which detection is in itself a difficult problem. Incomplete taxon sampling, or the extinction of the true donor lineage may render patterns difficult to interpret in a temporary fashion. Yet, for clades with no fossils this may be the only piece of information we have at hand, and the growing amount of sequence data is likely to minimize issues derived from incomplete sampling. The developed method, MaxTiC (for Maximal Time Consistency) [2], represents a very nice application of theoretical developments on the well-known « Feedback Arc Set » computer science problem to the evolutionary question of ordering nodes in a phylogeny. MaxTiC uses as input a species tree and a set of time constraints based on lateral gene transfers inferred using other softwares, and minimizes conflicts between node ordering and these time constraints. The application of MaxTiC on simulated datasets indicated that node ordering was fairly accurate [2]. MaxTiC is implemented in a freely available software, which represents original and relevant contribution to the field of evolutionary biology. References [1] Donoghue P and Smith M, editors. 2003. Telling the evolutionary time. CRC press. [2] Chauve C, Rafiey A, Davin AA, Scornavacca C, Veber P, Boussau B, Szöllősi GJ, Daubin V and Tannier E. 2017. MaxTiC: Fast ranking of a phylogenetic tree by Maximum Time Consistency with lateral gene transfers. bioRxiv 127548, ver. 6 of 6th November 2017. doi: 10.1101/127548 [3] Ropars J, Rodríguez de la Vega RC, Lopez-Villavicencio M, Gouzy J, Sallet E, Debuchy R, Dupont J, Branca A and Giraud T. 2015. Adaptive horizontal gene transfers between multiple cheese-associated fungi. Current Biology 19, 2562–2569. doi: 10.1016/j.cub.2015.08.025 [4] Novo M, Bigey F, Beyne E, Galeote V, Gavory F, Mallet S, Cambon B, Legras JL, Wincker P, Casaregola S and Dequin S. 2009. Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proceeding of the National Academy of Science USA, 106, 16333–16338. doi: 10.1073/pnas.0904673106 [5] Naranjo-Ortíz MA, Brock M, Brunke S, Hube B, Marcet-Houben M, Gabaldón T. 2016. Widespread inter- and intra-domain horizontal gene transfer of d-amino acid metabolism enzymes in Eukaryotes. Frontiers in Microbiology 7, 2001. doi: 10.3389/fmicb.2016.02001 [6] Alexander WG, Wisecaver JH, Rokas A, Hittinger CT. 2016. Horizontally acquired genes in early-diverging pathogenic fungi enable the use of host nucleosides and nucleotides. Proceeding of the National Academy of Science USA. 113, 4116–4121. doi: 10.1073/pnas.1517242113 [7] Marcet-Houben M, Gabaldón T. 2010. Acquisition of prokaryotic genes by fungal genomes. Trends in Genetics. 26, 5–8. doi: 10.1016/j.tig.2009.11.007 | MaxTiC: Fast ranking of a phylogenetic tree by Maximum Time Consistency with lateral gene transfers | Cédric Chauve, Akbar Rafiey, Adrian A. Davin, Celine Scornavacca, Philippe Veber, Bastien Boussau, Gergely J Szöllosi, Vincent Daubin, and Eric Tannier | Lateral gene transfers (LGTs) between ancient species contain information about the relative timing of species diversification. Specifically, the ancestors of a donor species must have existed before the descendants of the recipient species. Hence... | | Bioinformatics & Computational Biology, Evolutionary Dynamics, Genome Evolution, Life History, Molecular Evolution, Phylogenetics / Phylogenomics | Tatiana Giraud | 2017-06-28 13:40:52 | ||
06 Oct 2017

Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebratesCombining molecular information on chromatin organisation with eQTLs and evolutionary conservation provides strong candidates for the evolution of gene regulation in mammalian brainsRecommended by Marc Robinson-Rechavi based on reviews by Marc Robinson-Rechavi and Charles DankoIn this manuscript [1], Francisco J. Novo proposes candidate non-coding genomic elements regulating neurodevelopmental genes. What is very nice about this study is the way in which public molecular data, including physical interaction data, is used to leverage recent advances in our understanding to molecular mechanisms of gene regulation in an evolutionary context. More specifically, evolutionarily conserved non coding sequences are combined with enhancers from the FANTOM5 project, DNAse hypersensitive sites, chromatin segmentation, ChIP-seq of transcription factors and of p300, gene expression and eQTLs from GTEx, and physical interactions from several Hi-C datasets. The candidate regulatory regions thus identified are linked to candidate regulated genes, and the author shows their potential implication in brain development. While the results are focused on a small number of genes, this allows to verify features of these candidates in great detail. This study shows how functional genomics is increasingly allowing us to fulfill the promises of Evo-Devo: understanding the molecular mechanisms of conservation and differences in morphology. References [1] Novo, FJ. 2017. Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebrates. bioRxiv, 150482, ver. 4 of Sept 29th, 2017. doi: 10.1101/150482 | Evolutionary analysis of candidate non-coding elements regulating neurodevelopmental genes in vertebrates | Francisco J. Novo | <p>Many non-coding regulatory elements conserved in vertebrates regulate the expression of genes involved in development and play an important role in the evolution of morphology through the rewiring of developmental gene networks. Available biolo... | | Genome Evolution | Marc Robinson-Rechavi | Marc Robinson-Rechavi, Charles Danko | 2017-06-29 08:55:41 |




