Modularity of genes involved in local adaptation to climate despite physical linkage
Katie E. Lotterhos, Sam Yeaman, Jon Degner, Sally Aitken, Kathryn Hodgins
https://doi.org/10.1101/202481
Differential effect of genes in diverse environments, their role in local adaptation and the interference between genes that are physically linked
Recommended by Sebastian Ernesto Ramos-Onsins based on reviews by Tanja Pyhäjärvi and 1 anonymous reviewer
The genome of eukaryotic species is a complex structure that experience many different interactions within itself and with the surrounding environment. The genetic architecture of a phenotype (that is, the set of genetic elements affecting a trait of the organism) plays a fundamental role in understanding the adaptation process of a species to, for example, different climate environments, or to its interaction with other species. Thus, it is fundamental to study the different aspects of the genetic architecture of the species and its relationship with its surronding environment. Aspects such as modularity (the number of genetic units and the degree to which each unit is affecting a trait of the organism), pleiotropy (the number of different effects that a genetic unit can have on an organism) or linkage (the degree of association between the different genetic units) are essential to understand the genetic architecture and to interpret the effects of selection on the genome. Indeed, the knowledge of the different aspects of the genetic architecture could clarify whether genes are affected by multiple aspects of the environment or, on the contrary, are affected by only specific aspects [1,2].
The work performed by Lotterhos et al. [3] sought to understand the genetic architecture of the adaptation to different environments in lodgepole pine (Pinus contorta), considering as candidate SNPs those previously detected as a result of its extreme association patterns to different environmental variables or to extreme population differentiation. This consideration is very important because the study is only relevant if the studied markers are under the effect of selection. Otherwise, the genetic architecture of the adaptation to different environments would be masked by other (neutral) kind of associations that would be difficult to interpret [4,5]. In order to understand the relationship between genetic architecture and adaptation, it is relevant to detect the association networks of the candidate SNPs with climate variables (a way to measure modularity) and if these SNPs (and loci) are affected by single or multiple environments (a way to measure pleiotropy).
The authors used co-association networks, an innovative approach in this field, to analyse the interaction between the environmental information and the genetic polymorphism of each individual. This methodology is more appropriate than other multivariate methods - such as analysis based on principal components - because it is possible to cluster SNPs based on associations with similar environmental variables. In this sense, the co-association networks allowed to both study the genetic and physical linkage between different co-associations modules but also to compare two different models of evolution: a Modular environmental response architecture (specific genes are affected by specific aspects of the environment) or a Universal pleiotropic environmental response architecture (all genes are affected by all aspects of the environment). The representation of different correlations between allelic frequency and environmental factors (named galaxy biplots) are especially informative to understand the effect of the different clusters on specific aspects of the environment (for example, the co-association network ‘Aridity’ shows strong associations with hot/wet versus cold/dry environments).
The analysis performed by Lotterhos et al. [3], although it has some unavoidable limitations (e.g., only extreme candidate SNPs are selected, limiting the results to the stronger effects; the genetic and physical map is incomplete in this species), includes relevant results and also implements new methodologies in the field. To highlight some of them: the preponderance of a Modular environmental response architecture (evolution in separated modules), the detection of physical linkage among SNPs that are co-associated with different aspects of the environment (which was unexpected a priori), the implementation of co-association networks and galaxy biplots to see the effect of modularity and pleiotropy on different aspects of environment. Finally, this work contains remarkable introductory Figures and Tables explaining unambiguously the main concepts [6] included in this study. This work can be treated as a starting point for many other future studies in the field.
References
[1] Hancock AM, Brachi B, Faure N, Horton MW, Jarymowycz LB, Sperone FG, Toomajian C, Roux F & Bergelson J. 2011. Adaptation to climate across the Arabidopsis thaliana genome. Science 334: 83–86. doi: 10.1126/science.1209244
[2] Wagner GP & Zhang J. The pleiotropic structure of the genotypephenotype map: the evolvability of complex organisms. Nature Review Genetics 12: 204–213. doi: 10.1038/nrg2949
[3] Lotterhos KE, Yeaman S, Degner J, Aitken S, Hodgins K. 2018. Modularity of genes involved in local adaptation to climate despite physical linkage. bioRxiv 202481, ver. 4 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/202481
[4] Lotterhos KE & Whitlock MC. 2014. Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Molecular Ecology 23: 2178–2192. doi: 10.1111/mec.12725
[5] Lotterhos KE & Whitlock MC. 2015. The relative power of genome scans to detect local adaptation depends on sampling design and statistical method. Molecular Ecology 24: 1031–1046. doi: 10.1111/mec.13100
[6] Paaby AB & Rockman MV. 2013. The many faces of pleiotropy. Trends in Genetics 29: 66-73. doi: 10.1016/j.tig.2012.10.010
| Modularity of genes involved in local adaptation to climate despite physical linkage | Katie E. Lotterhos, Sam Yeaman, Jon Degner, Sally Aitken, Kathryn Hodgins | <p>Background: Physical linkage among genes shaped by different sources of selection is a fundamental aspect of genetic architecture. Theory predicts that evolution in complex environments selects for modular genetic architectures and high recombi... |  | Adaptation, Bioinformatics & Computational Biology, Genome Evolution | Sebastian Ernesto Ramos-Onsins | | 2017-10-15 19:21:57 | View |
The dynamics of preferential host switching: host phylogeny as a key predictor of parasite prevalence and distribution
Jan Engelstaedter & Nicole Fortuna
https://doi.org/10.1101/209254
Shift or stick? Untangling the signatures of biased host switching, and host-parasite co-speciation
Recommended by Lucy Weinert based on reviews by Damien de Vienne and Nathan Medd
Many emerging diseases arise by parasites switching to new host species, while other parasites seem to remain with same host lineage for very long periods of time, even over timescales where an ancestral host species splits into two or more new species. The ability to understand these dynamics would form an important part of our understanding of infectious disease.
Experiments are clearly important for understanding these processes, but so are comparative studies, investigating the variation that we find in nature. Such comparative data do show strong signs of non-randomness, and this suggests that the epidemiological and ecological processes might be predictable, at least in part. For example, when we map patterns of parasite presence/absence onto host phylogenies, we often find that certain host clades harbour many more parasites than expected, or that closely-related hosts harbour closely-related parasites. Nevertheless, it remains difficult to interpret these patterns to make inferences about ecological and epidemiological processes. This is partly because non-random associations can arise in multiple ways. For example, parasites might be inherited from the common ancestor of related hosts, or might switch to new hosts, but preferentially establish on novel hosts that are closely related to their existing host. Infection might also influence the shape of host phylogeny, either by increasing the rate of host extinction or, conversely, increasing the rate of speciation (as with manipulative symbionts that might induce reproductive isolation).
These various processes have, by and large, been studied in isolation, but the model introduced by Engelstädter and Fortuna [1], makes an important first step towards studying them together. Without such combined analyses, we will not be able to tell if the processes have their own unique signatures, or whether the same sort of non-randomness can arise in multiple ways.
A major finding of the work is that the size of a host clade can be an important determinant of its overall infection level. This had been shown in previous work, assuming that the host phylogeny was fixed, but the current paper shows that it extends also to situations where host extinction and speciation takes place at a comparable rate to host shifting. This finding, then, calls into question the natural assumption that a clade of host species that is highly parasite ridden, must have some genetic or ecological characteristic that makes them particularly prone to infection, arguing that the clade size, rather than any characteristic of the clade members, might be the important factor. It will be interesting to see whether this prediction about clade size is borne out with comparative studies.
Another feature of the study is that the framework is naturally extendable, to include further processes, such as the influence of parasite presence on extinction or speciation rates. No doubt extensions of this kind will form the basis of important future work.
References
[1] Engelstädter J and Fortuna NZ. 2018. The dynamics of preferential host switching: host phylogeny as a key predictor of parasite prevalence and distribution. bioRxiv 209254 ver. 5 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/209254
| The dynamics of preferential host switching: host phylogeny as a key predictor of parasite prevalence and distribution | Jan Engelstaedter & Nicole Fortuna | <p>New parasites commonly arise through host-shifts, where parasites from one host species jump to and become established in a new host species. There is much evidence that the probability of host-shifts decreases with increasing phylogenetic dist... | 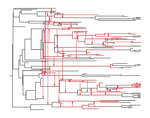 | Bioinformatics & Computational Biology, Evolutionary Epidemiology, Evolutionary Theory, Macroevolution, Phylogenetics / Phylogenomics, Species interactions | Lucy Weinert | | 2017-10-30 02:06:06 | View |
Variation in competitive ability with mating system, ploidy and range expansion in four Capsella species
Xuyue Yang, Martin Lascoux and Sylvain Glémin
https://doi.org/10.1101/214866
When ecology meets genetics: Towards an integrated understanding of mating system transitions and diversity
Recommended by Sylvain Billiard and Henrique Teotonio based on reviews by Yaniv Brandvain, Henrique Teotonio and 1 anonymous reviewer
In the 19th century, C. Darwin and F. Delpino engaged in a debate about the success of species with different reproduction modes, with the later favouring the idea that monoecious plants capable of autonomous selfing could spread more easily than dioecious plants (or self-incompatible hermaphroditic plants) if cross-pollination opportunities were limited [1]. Since then, debate has never faded about how natural selection is responsible for transitions to selfing and can explain the diversity and distribution of reproduction modes we observe in the natural world [2, 3].
Explanations for mating systems diversity, and transitions to selfing in particular, generally fall into two categories: either genetic or ecological. On the genetic side, many theoretical works showed a critical role for mutation load and inbreeding depression, transmission advantage and reproductive assurance in the evolution of selfing, e.g. [4]. Many experimental works were conducted to test theoretical hypotheses and predictions, especially regarding the magnitude of inbreeding depression; see [5] for a review. Ecologically, the presence of selfing populations is usually correlated with fragmented and harsh habitats, on the periphery of ancestral outcrossing populations. The cause of this distribution could be that selfers are better dispersers and colonizers than outcrossers, or variations in other life-history traits [6]. Yet, few experiments were run to assess whether selfing species or populations have effectively different ecological characteristics, and even scarcer are experiments evaluating both the roles of mutational load and life-history traits evolution. This is the aim of the present study by X. Yang et al [7].
The study of Yang et al [7], together with that of Petrone Mendoza et al. [8], supervised by S. Glémin and M. Lascoux, is probably one of the first to conduct experiments where the competitive abilities are compared between and within species. Using 4 species of the Capsella genus, annual plants from the mustard family, they tested the theoretical predictions that i) the transition from outcrossing to selfing resulted in reduced competitive ability at higher densities, because of the accumulation of deleterious mutations and/or the evolution of life-history traits in an open habitat and a colonization/dispersal trade-off; ii) that reduced competitive ability of selfers should be less pronounced in polyploid then diploid species because the effect of partially recessive deleterious mutations would be buffered; and iii) that competitive ability of selfers should decline with historical range expansion because of the expansion load [9].
Of the 4 Capsella species studied, only one of them, presumably the ancestral, is a diploid outcrosser with a small distribution but large population sizes. The three other species are selfers, two diploids with independent histories of transitions from outcrossing, and another, tetraploid, resulting from a recent hybridization between one of the diploid selfer and the diploid outcrossing ancestor. Many accessions from each species were sampled and individuals assayed for their competitive ability against a tester species or alone, for vegetative and reproductive traits. The measured vegetative traits (rosette surface at two stages, growth rate and flowering probability) showed no differentiation between selfers and outcrossers. To the contrary, reproductive traits (number of flowers) followed theoretical predictions: selfing species are more sensitive to competition than the outcrossing species, with polyploid selfing species being intermediate between the diploid selfers and the diploid outcrosser, and within the tetraploid selfing species (where sampling was quite significant across a large geographical range) sensitivity to competition increased with range expansion.
The study of Yang et al. [7] suffers from several limitations, such that alternative explanations cannot be discarded in the absence of further experimental data. They nonetheless provide the reader with a nice discussion and prospects on how to untwine the causes and the consequences of transitions to selfing. Their study also brings up to date questions about the joint evolution of mating system and life-history traits, which needs a renewed interest from an empirical and theoretical point of view. The results of Yang et al. raise for instance the question of whether it is indeed expected that only reproductive traits, and not vegetative traits, should evolve with the transition to selfing.
The recommandation and evaluation of this paper have been made in collaboration with Thomas Lesaffre.
References
[1] Darwin, C. R. (1876). The effects of cross and self fertilization in the vegetable kingdom. London: Murray.
[2] Stebbins, G. L. (1957). Self fertilization and population variability in the higher plants. The American Naturalist, 91, 337-354. doi: 10.1086/281999
[3] Harder, L.D. & Barrett, S. C. H. (2006). Ecology and evolution of flowers. Oxford: Oxford University Press.
[4] Porcher, E. & Lande, R. (2005). The evolution of self-fertilization and inbreeding depression under pollen discounting and pollen limitation. Journal of Evolutionary Biology, 18(3), 497-508. doi: 10.1111/j.1420-9101.2005.00905.x
[5] Winn, A.A., et al. (2011). Analysis of inbreeding depression in mixed-mating plants provides evidence for selective interference and stable mixed mating. Evolution, 65(12), 3339-3359. doi: 10.1111/j.1558-5646.2011.01462.x
[6] Munoz, F., Violle, C. & Cheptou, P.-O. (2016). CSR ecological strategies and plant mating systems: outcrossing increases with competitiveness but stress-tolerance is related to mixed mating. Oikos, 125(9), 1296-1303. doi: 10.1111/oik.02328
[7] Yang, X., Lascoux, M. & Glémin, S (2018). Variation in competitive ability with mating system, ploidy and range expansion in four Capsella species. bioRxiv, 214866, ver. 5 recommended and peer-reviewed by PCI Evol Biol. doi: 10.1101/214866
[8] Petrone Mendoza, S., Lascoux, M. & Glémin, S. (2018). Competitive ability of Capsella species with different mating systems and ploidy levels. Annals of Botany 121(6), 1257-1264. doi: 10.1093/aob/mcy014
[9] Peischl, S. & Excoffier, L. (2015). Expansion load: recessive mutations and the role of standing genetic variation. Molecular Ecology, 24(9): 2084-2094. doi: 10.1111/mec.13154
| Variation in competitive ability with mating system, ploidy and range expansion in four Capsella species | Xuyue Yang, Martin Lascoux and Sylvain Glémin | <p>Self-fertilization is often associated with ecological traits corresponding to the ruderal strategy in Grime’s Competitive-Stress-tolerant-Ruderal (CSR) classification of ecological strategies. Consequently, selfers are expected to be less comp... |  | Evolutionary Ecology, Population Genetics / Genomics, Reproduction and Sex, Species interactions | Sylvain Billiard | | 2017-11-06 19:54:52 | View |
A new approach to DNA-aided ancestral trait reconstruction in mammals
Recommended by Nicolas Galtier and Belinda Chang
Reconstructing ancestral character states is an exciting but difficult problem. The fossil record carries a great deal of information, but it is incomplete and not always easy to connect to data from modern species. Alternatively, ancestral states can be estimated by modelling trait evolution across a phylogeny, and fitting to values observed in extant species. This approach, however, is heavily dependent on the underlying assumptions, and typically results in wide confidence intervals.
An alternative approach is to gain information on ancestral character states from DNA sequence data. This can be done directly when the trait of interest is known to be determined by a single, or a small number, of major effect genes. In some of these cases it can even be possible to investigate an ancestral trait of interest by inferring and resurrecting ancestral sequences in the laboratory. Examples where this has been successfully used to address evolutionary questions range from the nocturnality of early mammals [1], to the loss of functional uricases in primates, leading to high rates of gout, obesity and hypertension in present day humans [2]. Another possibility is to rely on correlations between species traits and the genome average substitution rate/process. For instance, it is well established that the ratio of nonsynonymous to synonymous substitution rate, dN/dS, is generally higher in large than in small species of mammals, presumably due to a reduced effective population size in the former. By estimating ancestral dN/dS, one can therefore gain information on ancestral body mass (e.g. [3-4]).
The interesting paper by Wu et al. [5] further develops this second possibility of incorporating information on rate variation derived from genomic data in the estimation of ancestral traits. The authors analyse a large set of 1185 genes in 89 species of mammals, without any prior information on gene function. The substitution rate is estimated for each gene and each branch of the mammalian tree, and taken as an indicator of the selective constraint applying to a specific gene in a specific lineage – more constraint, slower evolution. Rate variation is modelled as resulting from a gene effect, a branch effect, and a gene X branch interaction effect, which captures lineage-specific peculiarities in the distribution of functional constraint across genes. The interaction term in terminal branches is regressed to observed trait values, and the relationship is used to predict ancestral traits from interaction terms in internal branches. The power and accuracy of the estimates are convincingly assessed via cross validation. Using this method, the authors were also able to use an unbiased approach to determine which genes were the main contributors to the evolution of the life-history traits they reconstructed.
The ancestors to current placental mammals are predicted to have been insectivorous - meaning that the estimated distribution of selective constraint across genes in basal branches of the tree resembles that of extant insectivorous taxa - consistent with the mainstream palaeontological hypothesis. Another interesting result is the prediction that only nocturnal lineages have passed the Cretaceous/Tertiary boundary, so that the ancestors of current orders of placentals would all have been nocturnal. This suggests that the so-called "nocturnal bottleneck hypothesis" should probably be amended. Similar reconstructions are achieved for seasonality, sociality and monogamy – with variable levels of uncertainty.
The beauty of the approach is to analyse the variance, not only the mean, of substitution rate across genes, and their methods allow for the identification of the genes contributing to trait evolution without relying on functional annotations. This paper only analyses discrete traits, but the framework can probably be extended to continuous traits as well.
References
[1] Bickelmann C, Morrow JM, Du J, Schott RK, van Hazel I, Lim S, Müller J, Chang BSW, 2015. The molecular origin and evolution of dim-light vision in mammals. Evolution 69: 2995-3003. doi: https://doi.org/10.1111/evo.12794
[2] Kratzer, JT, Lanaspa MA, Murphy MN, Cicerchi C, Graves CL, Tipton PA, Ortlund EA, Johnson RJ, Gaucher EA, 2014. Evolutionary history and metabolic insights of ancient mammalian uricases. Proceedings of the National Academy of Science, USA 111:3763-3768. doi: https://doi.org/10.1073/pnas.1320393111
[3] Lartillot N, Delsuc F. 2012. Joint reconstruction of divergence times and life-history evolution in placental mammals using a phylogenetic covariance model. Evolution 66:1773-1787. doi: https://doi.org/10.1111/j.1558-5646.2011.01558.x
[4] Romiguier J, Ranwez V, Douzery EJ, Galtier N. 2013. Genomic evidence for large, long-lived ancestors to placental mammals. Molecular Biology and Evolution 30:5-13. doi: https://doi.org/10.1093/molbev/mss211
[5] Wu J, Yonezawa T, Kishino H. 2016. Rates of Molecular Evolution Suggest Natural History of Life History Traits and a Post-K-Pg Nocturnal Bottleneck of Placentals. Current Biology 27: 3025-3033. doi: https://doi.org/10.1016/j.cub.2017.08.043
| Rates of Molecular Evolution Suggest Natural History of Life History Traits and a Post-K-Pg Nocturnal Bottleneck of Placentals | Wu J, Yonezawa T, Kishino H. | Life history and behavioral traits are often difficult to discern from the fossil record, but evolutionary rates of genes and their changes over time can be inferred from extant genomic data. Under the neutral theory, molecular evolutionary rate i... |  | Bioinformatics & Computational Biology, Life History, Molecular Evolution, Paleontology, Phylogenetics / Phylogenomics | Nicolas Galtier | | 2017-11-10 14:52:26 | View |
One (more) step towards a dynamic view of the Latitudinal Diversity Gradient
Recommended by Joaquín Hortal and Juan Arroyo based on reviews by Juan Arroyo, Joaquín Hortal, Arne Mooers, Joaquin Calatayud and 2 anonymous reviewers based on reviews by Juan Arroyo, Joaquín Hortal, Arne Mooers, Joaquin Calatayud and 2 anonymous reviewers
The Latitudinal Diversity Gradient (LDG) has fascinated natural historians, ecologists and evolutionary biologists ever since [1] described it about 200 years ago [2]. Despite such interest, agreement on the origin and nature of this gradient has been elusive. Several tens of hypotheses and models have been put forward as explanations for the LDG [2-3], that can be grouped in ecological, evolutionary and historical explanations [4] (see also [5]). These explanations can be reduced to no less than 26 hypotheses, which account for variations in ecological limits for the establishment of progressively larger assemblages, diversification rates, and time for species accumulation [5]. Besides that, although in general the tropics hold more species, different taxa show different shapes and rates of spatial variation [6], and a considerable number of groups show reverse patterns, with richer assemblages in cold temperate regions (see e.g. [7-9]).
Understanding such complexity needs integrating ecological and evolutionary research into the wide temporal and spatial perspectives provided by the burgeoning field of biogeography. This integrative discipline ¬–that traces back to Humboldt himself (e.g. [10])– seeks to put together historical and functional explanations to explain the complex dynamics of Earth’s biodiversity. Different to quantum physicists, biogeographers cannot pursue the ultimate principle behind the patterns we observe in nature due to the interplay of causes and effects, which in fact tell us that there is not such a single principle. Rather, they need to identify an array of basic principles coming from different perspectives, to then integrate them into models that provide realistic –but never simple– explanations to biodiversity gradients such as LDG (see, e.g., [5; 11]). That is, rather than searching for a sole explanation, research on the LDG must aim to identify as many signals hidden in the pattern as possible, and provide hypotheses or models that account for these signals. To later integrate them and, whenever possible, to validate them with empirical data on the organisms’ distribution, ecology and traits, phylogenies, fossils, etc.
Within this context, Meseguer & Condamine [12] provide a novel perspective to LDG research using phylogenetic and fossil evidence on the origin and extinction of taxa within the turtle, crocodile and lizard (i.e. squamate) lineages. By digging into deep time down to the Triassic (about 250 Myr ago) they are able to identify several episodes of flattening and steepening of the LDG for these three clades. Strikingly, their results show similar diversification rates in the northern hemisphere and in the equator during the over 100 Myr long global greenhouse period that extends from the late Jurassic to the Cretaceous and early Neogene. During this period, the LDG for these three groups would have appeared quite even across a mainly tropical Globe, although the equatorial regions were apparently much more evolutionarily dynamic. The equator shows much higher rates of origination and extinction of branches throughout the Cretaceous, but they counteract each other so net diversification is similar to that of the northern hemisphere in all three groups. The transition to a progressively colder Earth in the Paleogene (starting around 50 Myr ago) provokes a mass extinction in the three clades, which is compensated in the equator by the dispersal of many taxa from the areas that currently pertain to the Holarctic biogeographical realm. Finally, during the coldhouse Earth’s climatic conditions of the Neogene only squamates show significant positive diversification rates in extratropical areas, while the diversity of testudines remains, and crocodiles continue declining progressively towards oblivion in the whole world.
Meseguer & Condamine [12] attribute these temporal patterns to the so-called asymmetric gradient of extinction and dispersal (AGED) framework. Here, the dynamics of extinction-at and dispersal-from high latitudes during colder periods increase the steepness of the LDG. Whereas the gradient flattens when Earth warms up as a result of dispersal from the equator followed by increased diversification in extratropical regions. This idea in itself is not new, for the influence of climatic oscillations on diversification rates is well known, at least for the Pleistocene Ice Ages [13], as is the effect of niche conservatism on the LDG [14]. Nevertheless, Meseguer & Condamine’s AGED provides a synthetic verbal model that could allow integrating the three main types of processes behind the LDG into a single framework. To do this it would be necessary to combine AGED’s cycles of dispersal and diversification with realistic models of: (1) the ecological limits to host rich assemblages in the colder and less productive temperate climatic domains; (2) the variations in diversification rates with shifts in temperature and/or energy regimes; and (3) the geographical patterns of climatic oscillation through time that determine the time for species accumulation in each region.
Integrating these models may allow transposing Meseguer & Condamine’s [12] framework into the more mechanistic macroecological models advocated by Pontarp et al. [5]. This type of mechanistic models has been already used to understand the development of biodiversity gradients through the climatic oscillations of the Pleistocene and the Quaternary (e.g. [11]). So the challenge in this case would be to generate a realistic scenario of geographical dynamics that accounts for plate tectonics and long-term climatic oscillations. This is still a major gap and we would benefit from the integrated work by historical geologists and climatologists here. For instance, there is little doubt about the progressive cooling through the Cenozoic based in isotope recording in sea floor sediments [15]. Meseguer & Condamine [12] use this evidence for separating greenhouse, transition and coldhouse world scenarios, which should not be a problem for these rough classes. However, a detailed study of the evolutionary correlation of true climate variables across the tree of life is still pending, as temperature is inferred only for sea water in an ice-free ocean, say earlier half of the Cenozoic [15]. Precipitation regime is even less known. Such scenario would provide a scaffold upon which the temporal dynamics of several aspects of the generation and loss of biodiversity can be modelled. Additionally, one of the great advantages of selecting key clades to study the LDG would be to determine the functional basis of diversification. There are species traits that are well known to affect speciation and extinction probabilities, such as reproductive strategies or life histories (e.g. [16]). Whereas these traits might also be a somewhat redundant effect of climatic causes, they might foster (i.e. “extended reinforcement”, [17]) or slow diversification. Even so, it is unlikely that such a model would account for all the latitudinal variation in species richness. But it will at least provide a baseline for the main latitudinal variations in the diversity of the regional communities (sensu [18]) worldwide. Within this context the effects of recent ecological, evolutionary and historical processes, such as environmental heterogeneity, current diversification rates or glacial cycles, will only modify the general LDG pattern resulting from the main processes contained in Meseguer & Condamine’s AGED, thereby providing a more comprehensive understanding of the geographical gradients of diversity.
References
[1] Humboldt, A. v. (1808). Ansichten der Natur, mit wissenschaftlichen Erläuterungen. J. G. Cotta, Tübingen.
[2] Hawkins, B. A. (2001). Ecology's oldest pattern? Trends in Ecology & Evolution, 16, 470. doi: 10.1016/S0169-5347(01)02197-8
[3] Lomolino, M. V., Riddle, B. R. & Whittaker, R. J. (2017). Biogeography. Fifth Edition. Sinauer Associates, Inc., Sunderland, Massachussets.
[4] Mittelbach, G. G., Schemske, D. W., Cornell, H. V., Allen, A. P., Brown, J. M., Bush, M. B., Harrison, S. P., Hurlbert, A. H., Knowlton, N., Lessios, H. A., McCain, C. M., McCune, A. R., McDade, L. A., McPeek, M. A., Near, T. J., Price, T. D., Ricklefs, R. E., Roy, K., Sax, D. F., Schluter, D., Sobel, J. M. & Turelli, M. (2007). Evolution and the latitudinal diversity gradient: speciation, extinction and biogeography. Ecology Letters, 10, 315-331. doi: 10.1111/j.1461-0248.2007.01020.x
[5] Pontarp, M., Bunnefeld L., Cabral, J. S., Etienne, R. S., Fritz, S. A., Gillespie, R. Graham, C. H., Hagen, O., Hartig, F., Huang, S., Jansson, R., Maliet, O., Münkemüller, T., Pellissier, L., Rangel, T. F., Storch, D., Wiegand, T. & Hurlbert, A. H. (2019). The latitudinal diversity gradient: novel understanding through mechanistic eco-evolutionary models. Trends in ecology & evolution, 34, 211-223. doi: 10.1016/j.tree.2018.11.009
[6] Hillebrand, H. (2004). On the generality of the latitudinal diversity gradient. The American Naturalist, 163, 192-211. doi: 10.1086/381004
[7] Santos, A. M. C. & Quicke, D. L. J. (2011). Large-scale diversity patterns of parasitoid insects. Entomological Science, 14, 371-382. doi: 10.1111/j.1479-8298.2011.00481.x
[8] Morinière, J., Van Dam, M. H., Hawlitschek, O., Bergsten, J., Michat, M. C., Hendrich, L., Ribera, I., Toussaint, E. F. A. & Balke, M. (2016). Phylogenetic niche conservatism explains an inverse latitudinal diversity gradient in freshwater arthropods. Scientific Reports, 6, 26340. doi: 10.1038/srep26340
[9] Weiser, M. D., Swenson, N. G., Enquist, B. J., Michaletz, S. T., Waide, R. B., Zhou, J. & Kaspari, M. (2018). Taxonomic decomposition of the latitudinal gradient in species diversity of North American floras. Journal of Biogeography, 45, 418-428. doi: 10.1111/jbi.13131
[10] Humboldt, A. v. (1805). Essai sur la geographie des plantes; accompagné d'un tableau physique des régions equinoxiales. Levrault, Paris.
[11] Rangel, T. F., Edwards, N. R., Holden, P. B., Diniz-Filho, J. A. F., Gosling, W. D., Coelho, M. T. P., Cassemiro, F. A. S., Rahbek, C. & Colwell, R. K. (2018). Modeling the ecology and evolution of biodiversity: Biogeographical cradles, museums, and graves. Science, 361, eaar5452. doi: 10.1126/science.aar5452
[12] Meseguer, A. S. & Condamine, F. L. (2019). Ancient tropical extinctions contributed to the latitudinal diversity gradient. bioRxiv, 236646, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/236646
[13] Jansson, R., & Dynesius, M. (2002). The fate of clades in a world of recurrent climatic change: Milankovitch oscillations and evolution. Annual review of ecology and systematics, 33(1), 741-777. doi: 10.1146/annurev.ecolsys.33.010802.150520
[14] Wiens, J. J., & Donoghue, M. J. (2004). Historical biogeography, ecology and species richness. Trends in ecology & evolution, 19, 639-644. doi: 10.1016/j.tree.2004.09.011
[15] Zachos, J. C., Dickens, G. R., & Zeebe, R. E. (2008). An early Cenozoic perspective on greenhouse warming and carbon-cycle dynamics. Nature, 451, 279-283. doi: 10.1038/nature06588
[16] Zúñiga-Vega, J. J., Fuentes-G, J. A., Ossip-Drahos, A. G., & Martins, E. P. (2016). Repeated evolution of viviparity in phrynosomatid lizards constrained interspecific diversification in some life-history traits. Biology letters, 12, 20160653. doi: 10.1098/rsbl.2016.0653
[17] Butlin, R. K., & Smadja, C. M. (2018). Coupling, reinforcement, and speciation. The American Naturalist, 191, 155-172. doi: 10.1086/695136
[18] Ricklefs, R. E. (2015). Intrinsic dynamics of the regional community. Ecology letters, 18, 497-503. doi: 10.1111/ele.12431
| Ancient tropical extinctions contributed to the latitudinal diversity gradient | Andrea S. Meseguer, Fabien Condamine | <p>Biodiversity currently peaks at the equator, decreasing toward the poles. Growing fossil evidence suggest that this hump-shaped latitudinal diversity gradient (LDG) has not been persistent through time, with similar species diversity across lat... | 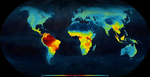 | Evolutionary Dynamics, Evolutionary Ecology, Macroevolution, Paleontology, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Joaquín Hortal | | 2017-12-20 14:58:01 | View |
Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands
Christelle Fraïsse, Anne Haguenauer, Karin Gerard, Alexandra Anh-Thu Weber, Nicolas Bierne, Anne Chenuil
https://doi.org/10.1101/239244
Introgression from related species reveals fine-scale structure in an isolated population of mussels and causes patterns of genetic-environment associations
Recommended by Marianne Elias based on reviews by Thomas Broquet and Tatiana Giraud
Assessing population connectivity is central to understanding population dynamics, and is therefore of great importance in evolutionary biology and conservation biology. In the marine realm, the apparent absence of physical barriers, large population sizes and high dispersal capacities of most organisms often result in no detectable structure, thereby hindering inferences of population connectivity. In a review paper, Gagnaire et al. [1] propose several ideas to improve detection of population connectivity. Notably, using simulations they show that under certain circumstances introgression from one species into another may reveal cryptic population structure within that second species.
The isolated Kerguelen archipelago in the south of Indian Ocean represents a typical situation where the structure of coastal marine organisms is expected to be difficult to detect. In an elegant genomic study, Fraïsse et al. [2] take advantage of introgression from foreign lineages to infer fine-grained population structure in a population of mussels around the Kerguelen archipelago, and investigate its association with environmental variables. Using a large panel of genome-wide markers (GBS) and applying a range of methods that unravel patterns of divergence and gene flow among lineages, they first find that the Kerguelen population is highly admixed, with a major genetic background corresponding to the southern mussel lineage Mytilus platensis introgressed by three northern lineages. By selecting a panel of loci enriched in ancestry-informative SNPs (ie, SNPs highly differentiated among northern lineages) they then detect a fine-scale genetic structure around the Kerguelen archipelago, and identify a major connectivity break. They further show an associating between the genetic structure and environmental variables, particularly the presence of Macrocystis kelp, a marker of habitat exposure to waves (a feature repeatedly evidenced to be important for mussels). While such association pattern could lead to the interpretation that differentiated SNPs correspond to loci directly under selection or linked with such loci, and even be considered as support for adaptive introgression, Fraïsse et al. [2] convincingly show by performing simulations that the genetic-environment association detected can be entirely explained by dispersal barriers associated with environmental variables (habitat-associated connectivity). They also explain why the association is better detected by ancestry-informative SNPs as predicted by Gagnaire et al. [1]. In addition, intrinsic genetic incompatibilities, which reduce gene flow, tend to become trapped at ecotones due to ecological selection, even when loci causing genetic incompatibilities are unlinked with loci involved in adaption to local ecological conditions (Bierne et al. [3]’s coupling hypothesis), leading to correlations between environmental variables and loci not involved in local adaptation. Notably, in Fraïsse et al. [2]’s study, the association between the kelp and ancestry-informative alleles is not consistent throughout the archipelago, casting further doubt on the implication of these alleles in local adaptation.
The study of Fraïsse et al. [2] is therefore an important contribution to evolutionary biology because 1) it provides an empirical demonstration that alleles of foreign origin can be pivotal to detect fine-scale connectivity patterns and 2) it represents a test case of Bierne et al. [3]’s coupling hypothesis, whereby introgressed alleles also enhance patterns of genetic-environment associations. Since genomic scan or GWAS approaches fail to clearly reveal loci involved in local adaptation, how can we disentangle environment-driven selection from intrinsic reproductive barriers and habitat-associated connectivity? A related question is whether we can reliably identify cases of adaptive introgression, which have increasingly been put forward as a mechanism involved in adaptation [4]. Unfortunately, there is no easy answer, and the safest way to go is to rely – where possible – on independent information [5], in particular functional studies of the detected loci, as is for example the case in the mimetic butterfly Heliconius literature (e. g., [6]) where several loci controlling colour pattern variation are well characterized.
References
[1] Gagnaire, P.-A., Broquet, T., Aurelle, D., Viard, F., Souissi, A., Bonhomme, F., Arnaud-Haond, S., & Bierne, N. (2015). Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evolutionary Applications, 8, 769–786. doi: 10.1111/eva.12288
[2] Fraïsse, C., Haguenauer, A., Gerard, K., Weber, A. A.-T., Bierne, N., & Chenuil, A. (2018). Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands. bioRxiv, 239244, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/239244
[3] Bierne, N., Welch, J., Loire, E., Bonhomme, F., & David, P. (2011). The coupling hypothesis: why genome scans may fail to map local adaptation genes. Molecular Ecology, 20, 2044–2072. doi: 10.1111/j.1365-294X.2011.05080.x
[4] Hedrick, P. W. (2013). Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Molecular Ecology, 22, 4606–4618. doi: 10.1111/mec.12415
[5] Ravinet, M., Faria, R., Butlin, R. K., Galindo, J., Bierne, N., Rafajlović, M., Noor, M. A. F., Mehlig, B., & Westram, A. M. (2017). Interpreting the genomic landscape of speciation: a road map for finding barriers to gene flow. Journal of Evolutionary Biology, 30, 1450–1477. doi: 10.1111/jeb.13047.
[6] Jay, P., Whibley, A., Frézal, L., Rodríguez de Cara, M. A., Nowell, R. W., Mallet, J., Dasmahapatra, K. K., & Joron, M. (2018). Supergene evolution triggered by the introgression of a chromosomal inversion. Current Biology, 28, 1839–1845.e3. doi: 10.1016/j.cub.2018.04.072
| Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands | Christelle Fraïsse, Anne Haguenauer, Karin Gerard, Alexandra Anh-Thu Weber, Nicolas Bierne, Anne Chenuil | <p>Reticulated evolution -i.e. secondary introgression / admixture between sister taxa- is increasingly recognized as playing a key role in structuring infra-specific genetic variation and revealing cryptic genetic connectivity patterns. When admi... |  | Hybridization / Introgression, Phylogeography & Biogeography, Population Genetics / Genomics | Marianne Elias | | 2017-12-28 14:16:16 | View |
Transgenerational cues about local mate competition affect offspring sex ratios in the spider mite Tetranychus urticae
Alison B. Duncan, Cassandra Marinosci, Céline Devaux, Sophie Lefèvre, Sara Magalhães, Joanne Griffin, Adeline Valente, Ophélie Ronce, Isabelle Olivieri
https://doi.org/10.1101/240127
Maternal effects in sex-ratio adjustment
Recommended by Dries Bonte based on reviews by 2 anonymous reviewers
Optimal sex ratios have been topic of extensive studies so far. Fisherian 1:1 proportions of males and females are known to be optimal in most (diploid) organisms, but many deviations from this golden rule are observed. These deviations not only attract a lot of attention from evolutionary biologists but also from population ecologists as they eventually determine long-term population growth. Because sex ratios are tightly linked to fitness, they can be under strong selection or plastic in response to changing demographic conditions. Hamilton [1] pointed out that an equality of the sex ratio breaks down when there is local competition for mates. Competition for mates can be considered as a special case of local resource competition. In short, this theory predicts females to adjust their offspring sex ratio conditional on cues indicating the level of local mate competition that their sons will experience. When cues indicate high levels of LMC mothers should invest more resources in the production of daughters to maximise their fitness, while offspring sex ratios should be closer to 50:50 when cues indicate low levels of LMC.
In isolated populations, Macke et al. [2] found sex ratio to evolve fast in response to changes in population sex-structure in the spider mite Tetranychus urticae. Spider mites are becoming top-models in evolutionary biology because of their easy housekeeping, fast generation times and well-studied genome [3]. The species is known to respond fast to changes in relatedness and kin-structure by changing its mating strategy [4], but also dispersal [5]. Sex ratio adjustments are likely mediated by differential investments in egg size, with small eggs possibly experiencing lower chances of fertilization, and thus to develop in haploid males [4].
Alison Duncan and colleagues [6] asked the question whether sex ratios change plastically in response to changes in the local population structure. They additionally questioned whether maternal effects could drive changes in sex-allocation of spider mite mothers. Indeed, theory predicts that if environmental changes are predictable across generations, intergenerational plasticity might be more adaptive than intragenerational plasticity [7]. Especially in spatially structured and highly dynamics populations, female spider mites may experience highly variable demographic conditions from one generation to another. During range expansions, spatial variation in local relatedness and inbreeding are documented to change and to impact eco-evolutionary trajectories as well (e.g. [8]).
Duncan et al. [6] specifically investigate whether the offspring sex ratio of T. urticae females changes in response to 1) the current number of females in the same patch, 2) the number of females in the patches of their mothers and 3) their relatedness to their mate. They surprisingly find the maternal environment to be more important than the actual experienced sex-ratio conditions. These insights thus show the maternal environment to be a reliable predictor of LMC experienced by grand-children. Maternal effects have been found to impact many traits, but this study is the first to convincingly demonstrate maternal effects in sex allocation. It therefore provides an alternative explanation of the apparent fast evolved responses under constant demographic conditions [2], and adds evidence to the importance of non-genetic trait changes for adaptation towards changing demographic and environmental conditions.
References
[1] Hamilton, W. D. (1967). Extraordinary Sex Ratios. Science, 156(3774), 477–488. doi: 10.1126/science.156.3774.477
[2] Macke, E., Magalhães, S., Bach, F., & Olivieri, I. (2011). Experimental evolution of reduced sex ratio adjustment under local mate competition. Science, 334(6059), 1127–1129. doi: 10.1126/science.1212177
[3] Grbić, M., Van Leeuwen, T., Clark, R. M., et al. (2011). The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature, 479(7374), 487–492. doi: 10.1038/nature10640
[4] Macke, E., Magalhães, S., Bach, F., & Olivieri, I. (2012). Sex-ratio adjustment in response to local mate competition is achieved through an alteration of egg size in a haplodiploid spider mite. Proceedings of the Royal Society B: Biological Sciences, 279(1747), 4634–4642. doi: 10.1098/rspb.2012.1598
[5] Bitume, E. V., Bonte, D., Ronce, O., Bach, F., Flaven, E., Olivieri, I., & Nieberding, C. M. (2013). Density and genetic relatedness increase dispersal distance in a subsocial organism. Ecology Letters, 16(4), 430–437. doi: 10.1111/ele.12057
[6] Duncan, A., Marinosci, C., Devaux, C., Lefèvre, S., Magalhães, S., Griffin, J., Valente, A., Ronce, O., Olivieri, I. (2018). Transgenerational cues about local mate competition affect offspring sex ratios in the spider mite Tetranychus urticae. BioRxiv, 240127, ver. 3. doi: 10.1101/240127
[7] Petegem, K. V., Moerman, F., Dahirel, M., Fronhofer, E. A., Vandegehuchte, M. L., Leeuwen, T. V., Wybouw, N., Stoks, R., Bonte, D. (2018). Kin competition accelerates experimental range expansion in an arthropod herbivore. Ecology Letters, 21(2), 225–234. doi: 10.1111/ele.12887
[8] Marshall, D. J., & Uller, T. (2007). When is a maternal effect adaptive? Oikos, 116(12), 1957–1963. doi: 10.1111/j.2007.0030-1299.16203.x
| Transgenerational cues about local mate competition affect offspring sex ratios in the spider mite Tetranychus urticae | Alison B. Duncan, Cassandra Marinosci, Céline Devaux, Sophie Lefèvre, Sara Magalhães, Joanne Griffin, Adeline Valente, Ophélie Ronce, Isabelle Olivieri | <p style="text-align: justify;">In structured populations, competition between closely related males for mates, termed Local Mate Competition (LMC), is expected to select for female-biased offspring sex ratios. However, the cues underlying sex all... |  | Evolutionary Ecology, Life History | Dries Bonte | | 2017-12-29 16:10:32 | View |
Range size dynamics can explain why evolutionarily age and diversification rate correlate with contemporary extinction risk in plants
Andrew J. Tanentzap, Javier Igea, Matthew G. Johnston, Matthew J. Larcombe
https://doi.org/10.1101/152215
Are both very young and the very old plant lineages at heightened risk of extinction?
Recommended by Arne Mooers based on reviews by Dan Greenberg and 1 anonymous reviewer
Human economic activity is responsible for the vast majority of ongoing extinction, but that does not mean lineages are being affected willy-nilly. For amphibians [1] and South African flowering plants [2], young species have a somewhat higher than expected chance of being threatened with extinction. In contrast, older Australian marsupial lineages seem to be more at risk [3]. Both of the former studies suggested that situations leading to peripheral isolation might simultaneously increase ongoing speciation and current threat via small geographic range, while the authors of the latter study suggested that older species might have evolved increasingly narrow niches. Here, Andrew Tanentzap and colleagues [4] dig deeper into the putative links between species age, niche breadth and threat status. Across 500-some plant genera worldwide, they find that, indeed, ""younger"" species (i.e. from younger and faster-diversifying genera) were more likely to be listed as imperiled by the IUCN, consistent with patterns for amphibians and African plants. Given this, results from their finer-level analyses of conifers are initially bemusing: here, ""older"" (i.e., on longer terminal branches) species were at higher risk. This would make conifers more like Australian marsupials, with the rest of the plants being more like amphibians. However, here where the data were more finely grained, the authors detected a second interesting pattern: using an intriguing matched-pair design, they detect a signal of conifer species niches seemingly shrinking as a function of age. The authors interpret this as consistent with increasing specialization, or loss of ancestral warm wet habitat, over paleontological time. It is true that conifers in general are older than plants more generally, with some species on branches that extend back many 10s of millions of years, and so a general loss of suitable habitat makes some sense. If so, both the pattern for all plants (small initial ranges heightening extinction) and the pattern for conifers (eventual increasing specialization or habitat contraction heightening extinction) could occur, each on a different time scale. As a coda, the authors detected no effect of age on threat status in palms; however, this may be both because palms have already lost species to climate-change induced extinction, and because they are thought to speciate more via long-distance dispersal and adaptive divergence then via peripheral isolation.
Given how quickly ranges can change, how hard it is to measure niche breadth, and the qualitatively different time scales governing past diversification and present-day extinction drivers, this is surely not the last word on the subject, even for plants. However, even the hint of a link between drivers of extinction in the Anthropocene and drivers of diversification through the ages is intellectually exciting and, perhaps, even, somehow, of practical importance.
References
[1] Greenberg, D. A., & Mooers, A. Ø. (2017). Linking speciation to extinction: Diversification raises contemporary extinction risk in amphibians. Evolution Letters, 1, 40–48. doi: 10.1002/evl3.4
[2] Davies, T. J., Smith, G. F., Bellstedt, D. U., Boatwright, J. S., Bytebier, B., Cowling, R. M., Forest, F., et al. (2011). Extinction risk and diversification are linked in a plant biodiversity hotspot. PLoS Biology, 9:e1000620. doi: 10.1371/journal.pbio.1000620
[3] Johnson, C. N., Delean S., & Balmford, A. (2002). Phylogeny and the selectivity of extinction in Australian marsupials. Animal Conservation, 5, 135–142. doi: 10.1017/S1367943002002196
[4] Tanentzap, A. J., Igea, J., Johnston, M. G., & Larcombe, M. G. (2018). Range size dynamics can explain why evolutionarily age and diversification rate correlate with contemporary extinction risk in plants. bioRxiv, 152215, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/152215
| Range size dynamics can explain why evolutionarily age and diversification rate correlate with contemporary extinction risk in plants | Andrew J. Tanentzap, Javier Igea, Matthew G. Johnston, Matthew J. Larcombe | <p>Extinction threatens many species, yet few factors predict this risk across the plant Tree of Life (ToL). Taxon age is one factor that may associate with extinction if occupancy of geographic and adaptive zones varies with time, but evidence fo... | 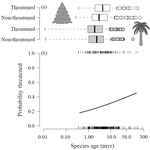 | Macroevolution, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Arne Mooers | | 2018-02-01 21:01:19 | View |
Field evidence for manipulation of mosquito host selection by the human malaria parasite, Plasmodium falciparum
Amelie Vantaux, Franck Yao, Domonbabele FdS Hien, Edwige Guissou, Bienvenue K Yameogo, Louis-Clement Gouagna, Didier Fontenille, Francois Renaud, Frederic Simard, Carlo Constantini, Frederic Thomas, Karine Mouline, Benjamin Roche, Anna Cohuet, Kounbobr R Dabire, Thierry Lefevre
https://doi.org/10.1101/207183
Malaria host manipulation increases probability of mosquitoes feeding on humans
Recommended by Alison Duncan based on reviews by Olivier Restif, Ricardo S. Ramiro and 1 anonymous reviewer
Parasites can manipulate their host’s behaviour to ensure their own transmission. These manipulated behaviours may be outside the range of ordinary host activities [1], or alter the crucial timing and/or location of a host’s regular activity. Vantaux et al show that the latter is true for the human malaria parasite, Plasmodium falciparum [2]. They demonstrate that three species of Anopheles mosquito were 24% more likely to choose human hosts, rather than other vertebrates, for their blood feed when they harboured transmissible stages (sporozoites) compared to when they were uninfected, or infected with non-transmissible malaria parasites [2]. Host choice is crucial for the malaria parasite Plasmodium falciparum to complete its life-cycle, as their host range is much narrower than the mosquito’s for feeding; P. falciparum can only develop in hominids, or closely related apes [3].
The study only shows this stage-dependent parasite manipulation retrospectively (by identifying host type and parasite stage in mosquitoes after their blood feed [2]). There was no difference in the preferences of infectious (with sporozoites) or un-infectious (infected without sporozoites, or uninfected) mosquitoes between human versus cow hosts in a choice test [2]. This suggests that the final decision about whether to feed occurs when the mosquito is in close range of the host.
This, coupled with previous findings, shows that vector manipulation is a fine-tuned business, that can act at multiple stages of the parasite life-cycle and on many behaviours [4]. Indeed, mosquitoes with non-transmissible Plasmodium stages (oocysts) are more reluctant to feed than sporozoite-infected mosquitoes [5] as vectors can be killed by their host whilst feeding, doing so before they are ready to transmit is risky for the malaria parasite. Thus, it seems that Plasmodium is, to some extent, master of its vector; commanding it not to feed when it cannot be transmitted, to feed when it is ready to be transmitted and to feed on the right type of host. What does this mean for our understanding of malaria transmission and epidemics?
Vantaux et al use a mathematical model, parameterised using data from this experiment, to highlight the consequences of this 24% increase in feeding on humans for P. falciparum transmission. They show that this increase raises the number of infectious bites humans receive from 4 (if sporozoite-infected mosquitoes had the same probability as uninfected mosquitoes) to 14 (an increase in 250%), for mosquitoes with a 15-day life-span, at ratios of 1:1 mosquitoes to humans. Longer mosquito life-spans and higher ratios of mosquitoes to humans further increases the number of infectious bites.
These results [2] have important implications for epidemiological forecasting and disease management. Public health strategies could focus on possible ways to trap sporozoite-infected mosquitoes, mimicking cues they use to locate their human hosts, or identify the behaviour of mosquitoes harbouring non-yet infectious Plasmodium, and trap them before they bite. Moreover, the results of the model show that failing to take into account the preference for humans of sporozoite-infected mosquitoes could underestimate the size of pending epidemics.
An important question previously raised is whether Plasmodium-induced alteration in host behaviour really is manipulation, or just a side-effect of being infected [4,5]. The fact that Vantaux et al show that these altered feeding behaviours increases the likelihood of transmission, in that a sporozoite-infected mosquito is more likely to feed on a human, strongly suggests that it is adaptive for the parasite [2]. Ultimately, to show that it is manipulation would require the identification of molecular factors released by Plasmodium that are responsible for physiological changes in the mosquito [6].
References
[1] Thomas, F., Schmidt-Rhaesa, A., Martin, G., Manu, C., Durand, P., & Renaud, F. (2002). Do hairworms (Nematomorpha) manipulate the water seeking behaviour of their terrestrial hosts? Journal of Evolutionary Biology, 15(3), 356–361. doi: 10.1046/j.1420-9101.2002.00410.x
[2] Vantaux, A., Yao, F., Hien, D. F., Guissou, E., Yameogo, B. K., Gouagna, L.-C., … Lefevre, T. (2018). Field evidence for manipulation of mosquito host selection by the human malaria parasite, Plasmodium falciparum. BioRxiv, 207183 ver 6. doi: 10.1101/207183
[3] Prugnolle, F., Durand, P., Ollomo, B., Duval, L., Ariey, F., Arnathau, C., … Renaud, F. (2011). A Fresh Look at the Origin of Plasmodium falciparum, the Most Malignant Malaria Agent. PLOS Pathogens, 7(2), e1001283. doi: 10.1371/journal.ppat.1001283
[4] Cator, L. J., Lynch, P. A., Read, A. F., & Thomas, M. B. (2012). Do malaria parasites manipulate mosquitoes? Trends in Parasitology, 28(11), 466–470. doi: 10.1016/j.pt.2012.08.004
[5] Cator, L. J., George, J., Blanford, S., Murdock, C. C., Baker, T. C., Read, A. F., & Thomas, M. B. (2013). “Manipulation” without the parasite: altered feeding behaviour of mosquitoes is not dependent on infection with malaria parasites. Proceedings. Biological Sciences, 280(1763), 20130711. doi: 10.1098/rspb.2013.0711
[6] Herbison, R., Lagrue, C., & Poulin, R. (2018). The missing link in parasite manipulation of host behaviour. Parasites & Vectors, 11. doi: 10.1186/s13071-018-2805-9
| Field evidence for manipulation of mosquito host selection by the human malaria parasite, Plasmodium falciparum | Amelie Vantaux, Franck Yao, Domonbabele FdS Hien, Edwige Guissou, Bienvenue K Yameogo, Louis-Clement Gouagna, Didier Fontenille, Francois Renaud, Frederic Simard, Carlo Constantini, Frederic Thomas, Karine Mouline, Benjamin Roche, Anna Cohuet, Kou... | <p>Whether the malaria parasite *Plasmodium falciparum* can manipulate mosquito host choice in ways that enhance parasite transmission toward human is unknown. We assessed the influence of *P. falciparum* on the blood-feeding behaviour of three of... |  | Evolutionary Ecology | Alison Duncan | | 2018-02-28 09:12:14 | View |
Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations
E. Noël, E. Fruitet, D. Lelaurin, N. Bonel, A. Ségard, V. Sarda, P. Jarne and P. David
https://doi.org//273367
Inbreeding compensates for reduced sexual selection in purging deleterious mutations
Recommended by Charles Baer based on reviews by 2 anonymous reviewers
Two evolutionary processes have been shown in theory to enhance the effects of natural selection in purging deleterious mutations from a population (here ""natural"" selection is defined as ""selection other than sexual selection""). First, inbreeding, especially self-fertilization, facilitates the removal of deleterious recessive alleles, the effects of which are largely hidden from selection in heterozygotes when mating is random. Second, sexual selection can facilitate the removal of deleterious alleles of arbitrary dominance, with little or no demographic cost, provided that deleterious effects are greater in males than in females (""genic capture""). Inbreeding (especially selfing) and sexual selection are often negatively correlated in nature. Empirical tests of the role of sexual selection in purging deleterious mutations have been inconsistent, potentially due to the positive relationship between sexual selection and intersexual genetic conflict.
In their preprint, Noël et al. [1] report a cleverly designed, and impressively long-term, experimental evolution study designed to tease apart the relative contributions of selfing and sexual selection in purging deleterious mutations, using the self-compatible hermaphroditic snail Physa acuta. Hermaphroditism relieves at least some of the potential conflict between males and females because each individual expresses traits of each sex. The authors report a 50-generation (ten years!) evolution experiment with four experimental treatments: Control (C), in which snails reproduced by mass mating (allowing sexual selection) and the next generation was sampled randomly from offspring in proportion to maternal family size; Male-selection (M) in which snails reproduced by mass mating but maternal family size was held constant, removing the opportunity for fertility selection; Female fertility selection (F) in which snails mated monogamously but fertility selection was imposed, and selfing (S), in which snails reproduced by selfing every other generation, alternating with monogamy + fertility selection. Juvenile survival was taken as the proxy for fitness and was measured for offspring of self-fertilization and of outcross matings. Each line type (C, M, F, S) was replicated twice.
The results are enviably clear-cut: after 50 generations of evolution, outcross fitness dropped precipitously in the F treatment (monogamy+female fertility selection) and remained at ancestral levels in the other three treatments. Clearly, sexual selection in males is more efficient at purging deleterious alleles than is female fertility selection. Similarly, inbreeding depression was reduced in the S lines relative to the other treatments, indicating that, unsurprisingly, deleterious recessive mutations of large effect are purged under strong inbreeding. Outcross fitness in the S lines did not decline, in contrast to the F lines, which indicates that deleterious mutations are on average slightly recessive.
Taken as a whole, this study by Noël et al. [1] provides a compelling empirical demonstration of the efficacy of both sexual selection and strong inbreeding as mechanisms of purging, and implicates sexual conflict as a potentially important factor in studies in which relaxation of sexual selection fails to result in purging.
References
[1] Noël, E., Fruitet, E., Lelaurin, D., Bonel, N., Segard, A., Sarda, V., Jarne, P., & David P. (2018). Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations. bioRxiv, 273367, ver. 3 recommended and peer-reviewed by PCI Evol Biol. doi: 10.1101/273367
| Sexual selection and inbreeding: two efficient ways to limit the accumulation of deleterious mutations | E. Noël, E. Fruitet, D. Lelaurin, N. Bonel, A. Ségard, V. Sarda, P. Jarne and P. David | <p>This preprint has been reviewed and recommended by Peer Community In Evolutionary Biology (https://dx.doi.org/10.24072/pci.evolbiol.100055). Theory and empirical data showed that two processes can boost selection against deleterious mutations, ... |  | Adaptation, Experimental Evolution, Reproduction and Sex, Sexual Selection | Charles Baer | Anonymous | 2018-03-01 08:12:37 | View |







