
Latest recommendations

| Id | Title▲ | Authors | Abstract | Picture | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
26 Oct 2021

Large-scale geographic survey provides insights into the colonization history of a major aphid pest on its cultivated apple host in Europe, North America and North AfricaThe evolutionary puzzle of the host-parasite-endosymbiont Russian doll for apples and aphidsRecommended by Ignacio Bravo based on reviews by Pedro Simões and 1 anonymous reviewerEach individual multicellular organism, each of our bodies, is a small universe. Every living surface -skin, cuticle, bark, mucosa- is the home place to milliards of bacteria, fungi and viruses. They constitute our microbiota. Some of them are essential for certain organisms. Other could not live without their hosts. For many species, the relationship between host and microbiota is so close that their histories are inseparable. The recognition of this biological inextricability has led to the notion of holobiont as the organism ensemble of host and microbiota. When individuals of a particular animal or plant species expand their geographical range, it is the holobiont that expands. And these processes of migration, expansion and colonization are often accompanied by evolutionary and ecological innovations in the interspecies relationships, at the macroscopic level (e.g. novel predator-prey or host-parasite interactions) and at the microscopic level (e.g. changes in the microbiota composition). From the human point of view, these novel interactions can be economically disastrous if they involve and threaten important crop or cattle species. And this is especially worrying in the present context of genetic standardization and intensification for mass-production on the one hand, and of climate change on the other. With this perspective, the international team led by Amandine Cornille presents a study aiming at understanding the evolutionary history of the rosy apple aphid Dysaphis plantaginea Passerini, a major pest of the cultivated apple tree Malus domestica Borkh (1). The apple tree was probably domesticated in Central Asia, and later disseminated by humans over the world in different waves, and it was probably introduced in Europe by the Greeks. It is however unclear when and where D. plantaginea started parasitizing the cultivated apple tree. The ancestral D. plantaginea could have already infected the wild ancestor of current cultivated apple trees, but the aphid is not common in Central Asia. Alternatively, it may have gained access only later to the plant, possibly via a host jump, from Pyrus to Malus that may have occurred in Asia Minor or in the Caucasus. In the present preprint, Olvera-Vázquez and coworkers have analysed over 650 D. plantaginea colonies from 52 orchards in 13 countries, in Western, Central and Eastern Europe as well as in Morocco and the USA. The authors have analysed the genetic diversity in the sampled aphids, and have characterized as well the composition of the associated endosymbiont bacteria. The analyses detect substantial recent admixture, but allow to identify aphid subpopulations slightly but significantly differentiated and isolated by distance, especially those in Morocco and the USA, as well as to determine the presence of significant gene flow. This process of colonization associated to gene flow is most likely indirectly driven by human interactions. Very interestingly, the data show that this genetic diversity in the aphids is not reflected by a corresponding diversity in the associated microbiota, largely dominated by a few Buchnera aphidicola variants. In order to determine polarity in the evolutionary history of the aphid-tree association, the authors have applied approximate Bayesian computing and machine learning approaches. Albeit promising, the results are not sufficiently robust to assess directionality nor to confidently assess the origin of the crop pest. Despite the large effort here communicated, the authors point to the lack of sufficient data (in terms of aphid isolates), especially originating from Central Asia. Such increased sampling will need to be implemented in the future in order to elucidate not only the origin and the demographic history of the interaction between the cultivated apple tree and the rosy apple aphid. This knowledge is needed to understand how this crop pest struggles with the different seasonal and geographical selection pressures while maintaining high genetic diversity, conspicuous gene flow, differentiated populations and low endosymbiontic diversity. References
| Large-scale geographic survey provides insights into the colonization history of a major aphid pest on its cultivated apple host in Europe, North America and North Africa | Olvera-Vazquez S.G., Remoué C., Venon A, Rousselet A., Grandcolas O., Azrine M., Momont L., Galan M., Benoit L., David G., Alhmedi A., Beliën T., Alins G., Franck P., Haddioui A., Jacobsen S.K., Andreev R., Simon S., Sigsgaard L., Guibert E., Tour... | <p style="text-align: justify;">With frequent host shifts involving the colonization of new hosts across large geographical ranges, crop pests are good models for examining the mechanisms of rapid colonization. The microbial partners of pest insec... | | Phylogeography & Biogeography, Population Genetics / Genomics, Species interactions | Ignacio Bravo | 2020-12-11 19:22:54 | ||
02 Jan 2019
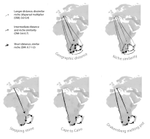
Leaps and bounds: geographical and ecological distance constrained the colonisation of the Afrotemperate by EricaThe colonization history of largely isolated habitatsRecommended by Andrea S. Meseguer based on reviews by Simon Joly, Florian Boucher and 2 anonymous reviewersThe build-up of biodiversity is the result of in situ speciation and immigration, with the interplay between geographical distance and ecological suitability determining the probability of an organism to establish in a new area. The relative contribution of these factors have long interested biogeographers, in particular to explain the distribution of organisms adapted to habitats that remained largely isolated, such as the colonization of oceanic islands or land waters. The focus of this study is the formation of the afrotemperate flora; patches of temperate vegetation separated by thousands of kilometers in Africa, with high levels of endemism described in the Cape region, the Drakensberg range and the high mountains of tropical east Africa [1]. The floristic affinities between these centers of endemism have frequently been explored but the origin of many afrotemperate lineages remains enigmatic [2]. References [1] Linder, H.P. 1990. On the relationship between the vegetation and floras of the Afromontane and the Cape regions of Africa. Mitteilungen aus dem Institut für Allgemeine Botanik Hamburg 23b:777–790. | Leaps and bounds: geographical and ecological distance constrained the colonisation of the Afrotemperate by Erica | Michael D. Pirie, Martha Kandziora, Nicolai M. Nuerk, Nicholas C. Le Maitre, Ana Laura Mugrabi de Kuppler, Berit Gehrke, Edward G.H. Oliver, and Dirk U. Bellstedt | <p>The coincidence of long distance dispersal and biome shift is assumed to be the result of a multifaceted interplay between geographical distance and ecological suitability of source and sink areas. Here, we test the influence of these factors o... | | Phylogeography & Biogeography | Andrea S. Meseguer | 2018-04-09 10:10:04 | ||
11 Sep 2017

POSTPRINT
Less effective selection leads to larger genomesColonisation of subterranean ecosystems leads to larger genome in waterlouse (Aselloidea)Recommended by Benoit Nabholz and Jochen B. W. WolfThe total amount of DNA utilized to store hereditary information varies immensely among eukaryotic organisms. Single copy genome sizes – disregarding differences due to ploidy - differ by more than three orders of magnitude ranging from a few million nucleotides (Mb) to hundreds of billions (Gb). With the ever-increasing availability of fully sequenced genomes we now know that most of the difference is due either to whole genome duplication or to variation in the abundance of repetitive elements. Regarding repetitive elements, the evolutionary forces underlying the large variation 'allowing' more or less elements in a genome remain largely elusive. A tentative correlation between an organism's complexity (however this may be adequately measured) and genome size, the so called C-value paradox [1], has long been dismissed. Studies testing for selection on secondary phenotypic effects associated with genome size (cell size, metabolic rates, nutrient availability) have yielded mixed results. Nonadaptive theories capitalizing on a role of deleterious insertion-deletion mutations and genetic drift as the main drivers have likewise received mixed support [2-3]. Overall, most evidence was derived from analyses across broad taxonomical scales [4-6]. Lefébure and colleagues [7] take a different approach. They confine their considerations to a homogeneous, restricted taxonomical group, isopod crustaceans of the superfamily Aselloidea. This taxonomic focus allows the authors to circumvent many of the confounding factors such as phylogenetic inertia, life history divergence and mutation rate variation that tend to trouble analyses across broad taxonomic timescales. Another important feature of the chosen system is the evolutionary independent transition of habitat use that has occurred at least 11 times. One group of species inhabits subterranean ecosystems (groundwater), another group thrives on surface water. Populations of the former live in low-energy habitats and are expected to be outnumbered by their surface dwelling relatives. Interestingly – and a precondition for the study - the groundwater species have significantly larger genomes (up to 137%). With this unique set-up, the authors are able to investigate the link between genome size and evolutionary forces related to a proxy of long-term population size by removing many of the confounding factors a priori. Upfront, we learn that the dN/dS ratio is higher in the groundwater species. This may either suggest prevalent positive selection or lower efficacy of purifying selection (relaxed constraint) in the group of species in which population sizes are expected to be low. Using a series of population genetic analyses the authors provide compelling evidence for the latter. Analyses are carefully conducted and include models for estimating the intensity and frequency of purifying and positive selection, the DoS (direction of selection) and α statistic. Next the authors also exclude the possibility that increased dN/dS of the subterranean groundwater species may be due to nonfunctionalization, which may result from the subterranean lifestyle. Overall, these analyses suggest relaxed constraint in smaller populations as the most plausible alternative to explain increased dN/dS ratios. In addition to the efficacy of selection, the authors estimate the timing of the ecological transition under the rationale that the amount of time a species may have been exposed to the subterranean habitat may reflect long term population sizes. To calibrate the 'colonization clock' they apply a neat trick based on the degree of degeneration of the opsin gene (as vision tends to get lost in these habitats). When finally testing which parameters may explain differences in genome size all factors – ecological status, selection efficiency as measured by dN/dS and colonization time - turned out to be significant predictors. Direct estimates of the short term effective population size Ne from polymorphism data, however, did not correlate with genome size. Ruling out the effect of other co-variates such as body size and growth rate the authors conclude that genome size was overall best predicted by long-term population size change upon habitat shift. In that the authors provide convincing evidence that the increase in genome size is linked to a decrease in long-term reduction of selection efficiency of subterranean species. Assuming a bias for insertion mutations over deletion mutations (which is usually the case in eukaryotes) this result is in agreement with the theory of mutational hazard [4-6]. This theory proposed by Michael Lynch postulates that the accumulation of non-functional DNA has a weak deleterious effect that can only be efficiently opposed by natural selection in species with high Ne. In conclusion, Lefébure and colleagues provide novel and welcome evidence supporting a 'neutralist' hypothesis of genome size evolution without the need to invoke an adaptive component. Methodologically, the study cautions against the common use of polymorphism-based estimates of Ne which are often obfuscated by transitory demographic change. Instead, alternative measures of selection efficacy linked to long-term population size may serve as better predictors of genome size. We hope that this study will stimulate additional work testing the link between Ne and genome size variation in other taxonomical groups [8-9]. Using genome sequences instead of the transcriptome approach applied here may concomitantly further our understanding of the molecular mechanisms underlying genome size change. References [1] Thomas, CA Jr. 1971. The genetic organization of chromosomes. Annual Review of Genetics 5: 237–256. doi: 10.1146/annurev.ge.05.120171.001321 [2] Ågren JA, Greiner S, Johnson MTJ, Wright SI. 2015. No evidence that sex and transposable elements drive genome size variation in evening primroses. Evolution 69: 1053–1062. doi: 10.1111/evo.12627 [3] Bast J, Schaefer I, Schwander T, Maraun M, Scheu S, Kraaijeveld K. 2016. No accumulation of transposable elements in asexual arthropods. Molecular Biology and Evolution 33: 697–706. doi: 10.1093/molbev/msv261 [4] Lynch M. 2007. The Origins of Genome Architecture. Sinauer Associates. [5] Lynch M, Bobay LM, Catania F, Gout JF, Rho M. 2011. The repatterning of eukaryotic genomes by random genetic drift. Annual Review of Genomics and Human Genetics 12: 347–366. doi: 10.1146/annurev-genom-082410-101412 [6] Lynch M, Conery JS. 2003. The origins of genome complexity. Science 302: 1401–1404. doi: 10.1126/science.1089370 [7] Lefébure T, Morvan C, Malard F, François C, Konecny-Dupré L, Guéguen L, Weiss-Gayet M, Seguin-Orlando A, Ermini L, Der Sarkissian C, Charrier NP, Eme D, Mermillod-Blondin F, Duret L, Vieira C, Orlando L, and Douady CJ. 2017. Less effective selection leads to larger genomes. Genome Research 27: 1016-1028. doi: 10.1101/gr.212589.116 [8] Lower SS, Johnston JS, Stanger-Hall KF, Hjelmen CE, Hanrahan SJ, Korunes K, Hall D. 2017. Genome size in North American fireflies: Substantial variation likely driven by neutral processes. Genome Biolology and Evolution 9: 1499–1512. doi: 10.1093/gbe/evx097 [9] Sessegolo C, Burlet N, Haudry A. 2016. Strong phylogenetic inertia on genome size and transposable element content among 26 species of flies. Biology Letters 12: 20160407. doi: 10.1098/rsbl.2016.0407 | Less effective selection leads to larger genomes | Tristan Lefébure, Claire Morvan, Florian Malard, Clémentine François, Lara Konecny-Dupré, Laurent Guéguen, Michèle Weiss-Gayet, Andaine Seguin-Orlando, Luca Ermini, Clio Der Sarkissian, N. Pierre Charrier, David Eme, Florian Mermillod-Blondin, Lau... | The evolutionary origin of the striking genome size variations found in eukaryotes remains enigmatic. The effective size of populations, by controlling selection efficacy, is expected to be a key parameter underlying genome size evolution. However... | | Evolutionary Theory, Genome Evolution, Molecular Evolution, Population Genetics / Genomics | Benoit Nabholz | 2017-09-08 09:39:23 | ||
15 Dec 2016

POSTPRINT
Limiting opportunities for cheating stabilizes virulence in insect parasitic nematodesApplication of kin theory to long-standing problem in nematode production for biocontrolRecommended by Thomas Sappington and Ruth Arabelle HufbauerMuch research effort has been extended toward developing systems for managing soil inhabiting insect pests of crops with entomopathogenic nematodes as biocontrol agents. Although small plot or laboratory experiments may suggest a particular insect pest is vulnerable to management in this way, it is often difficult to scale-up nematode production for application at the field- and farm scale to make such a tactic viable. Part of the problem is that entomopathogenic nematode strains must be propagated by serial passage in vivo, because storage by freezing decreases fitness. At the same time, serial propagation results in loss of virulence (ability to infect) over generations in the laboratory, a phenomenon called attenuation. To probe the underlying reasons for development of attenuation, as a prerequisite to designing strategies to mitigate it, Shapiro-Ilan and Raymond [1] turned to evolutionary theory of social conflict as a possible explanatory framework. Virulence of entomopathogenic nematodes depends on a combination of virulence factors, like various proteases, secreted by both the nematode and symbiotic bacteria to overcome host defenses. Attenuation is characterized in part by a reduced production of these factors. Invasion of a host involves simultaneous attack by a group of nematodes ("cooperators"), which together neutralize host defenses enough to allow individuals to successfully invade. "Cheaters" in the invading population can avoid the metabolic costs of producing virulence factors while reaping the benefits of infecting the host made vulnerable by the cooperators in the population. The authors hypothesize that an increase in frequency of cheaters may contribute to attenuation of virulence during serial propagation in the laboratory. The evolutionary dynamics of cheater frequency in a population have been explored in many contexts as part of kin selection theory. Cheaters can increase in a population by outcompeting cooperators in a host if overall relatedness within the invading population is low. Conversely, frequency of altruism, or costly cooperation, increases in a population if relatedness is high, which is enhanced by low effective dispersal. However, a population that is too isolated can suffer from inbreeding effects, and competition will occur mainly among relatives, which decreases the fitness benefits of altruism. Shapiro-Ilan and Raymond [1] tested changes in virulence and reproductive output in a serially propagated entomopathogenic nematode, Heterorhabditis floridensis. They compared lines of high or low relatedness, manipulated via multiplicity of infection (MOI) rates (where a low dose of nematodes gives high relatedness and a high dose gives low relatedness); and under global or local competition, manipulated by pooling populations emerging from all or only two host cadavers per generation, respectively. As predicted, treatments of high relatedness (low MOI) and global competition had the greatest level of reproduction, while all lines of low relatedness (high MOI) evolved decreased reproduction and decreased virulence, which led to extinction. The key finding was that lines in the high relatedness (low MOI) and low (local) competition treatment exhibited the most stable virulence through the 12 generations tested. Thus, to minimize attenuation of virulence while maintaining fitness of recently isolated entomopathogenic nematodes, the authors recommend insect hosts be inoculated with low doses of nematodes from inocula pools from as few cadavers as possible. The application of evolutionary theory, with a clever experimental design, to an important problem in pest management makes this paper particularly noteworthy. Reference [1] Shapiro-Ilan D, Raymond B. 2016. Limiting opportunities for cheating stabilizes virulence in insect parasitic nematodes. Evolutionary Applications 9:462-470. doi: 10.1111/eva.12348 | Limiting opportunities for cheating stabilizes virulence in insect parasitic nematodes | Shapiro-Ilan D. and B. Raymond | Cooperative secretion of virulence factors by pathogens can lead to social conflict when cheating mutants exploit collective secretion, but do not contribute to it. If cheats outcompete cooperators within hosts, this can cause loss of virulence.... | | Adaptation, Behavior & Social Evolution, Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology, Evolutionary Epidemiology, Evolutionary Theory, Experimental Evolution, Population Genetics / Genomics, Reproduction and Sex | Thomas Sappington | 2016-12-15 18:33:39 | ||
12 Nov 2020
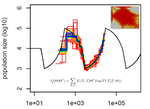
Limits and Convergence properties of the Sequentially Markovian CoalescentReview and Assessment of Performance of Genomic Inference Methods based on the Sequentially Markovian CoalescentRecommended by Stephan Schiffels based on reviews by 3 anonymous reviewers based on reviews by 3 anonymous reviewers
The human genome not only encodes for biological functions and for what makes us human, it also encodes the population history of our ancestors. Changes in past population sizes, for example, affect the distribution of times to the most recent common ancestor (tMRCA) of genomic segments, which in turn can be inferred by sophisticated modelling along the genome. References [1] Li, H., and Durbin, R. (2011). Inference of human population history from individual whole-genome sequences. Nature, 475(7357), 493-496. doi: https://doi.org/10.1038/nature10231 | Limits and Convergence properties of the Sequentially Markovian Coalescent | Thibaut Sellinger, Diala Abu Awad, Aurélien Tellier | <p>Many methods based on the Sequentially Markovian Coalescent (SMC) have been and are being developed. These methods make use of genome sequence data to uncover population demographic history. More recently, new methods have extended the original... | | Population Genetics / Genomics | Stephan Schiffels | Anonymous | 2020-07-25 10:54:48 | |
06 Sep 2022

Masculinization of the X-chromosome in aphid soma and gonadsSex-biased gene expression is not tissue-specific in Pea AphidsRecommended by Charles Baer and Tanja Schwander based on reviews by Ann Kathrin Huylmans and 1 anonymous reviewerSexual antagonism (SA), wherein the fitness interests of the sexes do not align, is inherent to organisms with two (or more) sexes. SA leads to intra-locus sexual conflict, where an allele that confers higher fitness in one sex reduces fitness in the other [1, 2]. This situation leads to what has been referred to as "gender load", resulting from the segregation of SA alleles in the population. Gender load can be reduced by the evolution of sex-specific (or sex-biased) gene expression. A specific prediction is that gene-duplication can lead to sub- or neo-functionalization, in which case the two duplicates partition the function in the different sexes. The conditions for invasion by a SA allele differ between sex-chromosomes and autosomes, leading to the prediction that (in XY or XO systems) the X should accumulate recessive male-favored alleles and dominant female-favored alleles; similar considerations apply in ZW systems ([3, but see 4]. Aphids present an interesting special case, for several reasons: they have XO sex-determination, and three distinct reproductive morphs (sexual females, parthenogenetic females, and males). Previous theoretical work by the lead author predict that the X should be optimized for male function, which was borne out by whole-animal transcriptome analysis [5]. Here [6], the authors extend that work to investigate “tissue”-specific (heads, legs and gonads), sex-specific gene expression. They argue that, if intra-locus SA is the primary driver of sex-biased gene expression, it should be generally true in all tissues. They set up as an alternative the possibility that sex-biased gene expression could also be driven by dosage compensation. They cite references supporting their argument that "dosage compensation (could be) stronger in the brain", although the underlying motivation for that argument appears to be based on empirical evidence rather than theoretical predictions. At any rate, the results are clear: all tissues investigated show masculinization of the X. Further, X-linked copies of gene duplicates were more frequently male-biased than duplicated autosomal genes or X-linked single-copy genes. To sum up, this is a nice empirical study with clearly interpretable (and interpreted) results, the most obvious of which is the greater sex-biased expression in sexually-dimorphic tissues. Unfortunately, as the authors emphasize, there is no general theory by which SA, variable dosage-compensation, and meiotic sex chromosome inactivation can be integrated in a predictive framework. It is to be hoped that empirical studies such as this one will motivate deeper and more general theoretical investigations. References [1] Rice WR, Chippindale AK (2001) Intersexual ontogenetic conflict. Journal of Evolutionary Biology 14: 685-693. https://doi.org/10.1046/j.1420-9101.2001.00319.x [2] Bonduriansky R, Chenoweth SF (2009) Intralocus sexual conflict. Trends Ecol Evol 24: 280-288. https://doi.org/10.1016/j.tree.2008.12.005 [3] Rice WR. (1984) Sex chromosomes and the evolution of sexual dimorphism. Evolution 38: 735-742. https://doi.org/10.1086/595754 [4] Fry JD (2010) The genomic location of sexually antagonistic variation: some cautionary comments. Evolution 64: 1510-1516. https://doi.org/10.1111%2Fj.1558-5646.2009.00898.x [5] Jaquiéry J, Rispe C, Roze D, Legeai F, Le Trionnaire G, Stoeckel S, et al. (2013) Masculinization of the X Chromosome in the Pea Aphid. PLoS Genetics 9. https://doi.org/10.1371/journal.pgen.1003690 [6] Jaquiéry J, Simon J-C, Robin S, Richard G, Peccoud J, Boulain H, Legeai F, Tanguy S, Prunier-Leterme N, Le Trionnaire G (2022) Masculinization of the X-chromosome in aphid soma and gonads. bioRxiv, 2021.08.13.453080, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.08.13.453080 | Masculinization of the X-chromosome in aphid soma and gonads | Julie Jaquiery, Jean-Christophe Simon, Stephanie Robin, Gautier Richard, Jean Peccoud, Helene Boulain, Fabrice Legeai, Sylvie Tanguy, Nathalie Prunier-Leterme, Gael Letrionnaire | <p>Males and females share essentially the same genome but differ in their optimal values for many phenotypic traits, which can result in intra-locus conflict between the sexes. Aphids display XX/X0 sex chromosomes and combine unusual X chromosome... | | Genetic conflicts, Genome Evolution, Reproduction and Sex | Charles Baer | 2021-08-16 08:56:08 | ||
07 Nov 2017

MaxTiC: Fast ranking of a phylogenetic tree by Maximum Time Consistency with lateral gene transfersDating nodes in a phylogeny using inferred horizontal gene transfersRecommended by Tatiana Giraud and Toni Gabaldon based on reviews by Alexandros Stamatakis, Mukul Bansal and 2 anonymous reviewersDating nodes in a phylogeny is an important problem in evolution and is typically performed by using molecular clocks and fossil age estimates [1]. The manuscript by Chauve et al. [2] reports a novel method, which uses lateral gene transfers to help ordering nodes in a species tree. The idea is that a lateral gene transfer can only occur between two species living at the same time, which indirectly informs on node relative ages in a phylogeny: the donor species cannot be more recent than the recipient species. Horizontal gene transfers are increasingly recognized as frequent, even in eukaryotes, and especially in micro-organisms that have little fossil records [3-7]. Yet, such an important source of information has been very rarely used so far for inferring relative node ages in phylogenies. In this context, the method by Chauve et al. [2] represents an innovative and original approach to a difficult problem. An obvious limitation of the approach is that it relies on inferences of horizontal transfers, which detection is in itself a difficult problem. Incomplete taxon sampling, or the extinction of the true donor lineage may render patterns difficult to interpret in a temporary fashion. Yet, for clades with no fossils this may be the only piece of information we have at hand, and the growing amount of sequence data is likely to minimize issues derived from incomplete sampling. The developed method, MaxTiC (for Maximal Time Consistency) [2], represents a very nice application of theoretical developments on the well-known « Feedback Arc Set » computer science problem to the evolutionary question of ordering nodes in a phylogeny. MaxTiC uses as input a species tree and a set of time constraints based on lateral gene transfers inferred using other softwares, and minimizes conflicts between node ordering and these time constraints. The application of MaxTiC on simulated datasets indicated that node ordering was fairly accurate [2]. MaxTiC is implemented in a freely available software, which represents original and relevant contribution to the field of evolutionary biology. References [1] Donoghue P and Smith M, editors. 2003. Telling the evolutionary time. CRC press. [2] Chauve C, Rafiey A, Davin AA, Scornavacca C, Veber P, Boussau B, Szöllősi GJ, Daubin V and Tannier E. 2017. MaxTiC: Fast ranking of a phylogenetic tree by Maximum Time Consistency with lateral gene transfers. bioRxiv 127548, ver. 6 of 6th November 2017. doi: 10.1101/127548 [3] Ropars J, Rodríguez de la Vega RC, Lopez-Villavicencio M, Gouzy J, Sallet E, Debuchy R, Dupont J, Branca A and Giraud T. 2015. Adaptive horizontal gene transfers between multiple cheese-associated fungi. Current Biology 19, 2562–2569. doi: 10.1016/j.cub.2015.08.025 [4] Novo M, Bigey F, Beyne E, Galeote V, Gavory F, Mallet S, Cambon B, Legras JL, Wincker P, Casaregola S and Dequin S. 2009. Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proceeding of the National Academy of Science USA, 106, 16333–16338. doi: 10.1073/pnas.0904673106 [5] Naranjo-Ortíz MA, Brock M, Brunke S, Hube B, Marcet-Houben M, Gabaldón T. 2016. Widespread inter- and intra-domain horizontal gene transfer of d-amino acid metabolism enzymes in Eukaryotes. Frontiers in Microbiology 7, 2001. doi: 10.3389/fmicb.2016.02001 [6] Alexander WG, Wisecaver JH, Rokas A, Hittinger CT. 2016. Horizontally acquired genes in early-diverging pathogenic fungi enable the use of host nucleosides and nucleotides. Proceeding of the National Academy of Science USA. 113, 4116–4121. doi: 10.1073/pnas.1517242113 [7] Marcet-Houben M, Gabaldón T. 2010. Acquisition of prokaryotic genes by fungal genomes. Trends in Genetics. 26, 5–8. doi: 10.1016/j.tig.2009.11.007 | MaxTiC: Fast ranking of a phylogenetic tree by Maximum Time Consistency with lateral gene transfers | Cédric Chauve, Akbar Rafiey, Adrian A. Davin, Celine Scornavacca, Philippe Veber, Bastien Boussau, Gergely J Szöllosi, Vincent Daubin, and Eric Tannier | Lateral gene transfers (LGTs) between ancient species contain information about the relative timing of species diversification. Specifically, the ancestors of a donor species must have existed before the descendants of the recipient species. Hence... | | Bioinformatics & Computational Biology, Evolutionary Dynamics, Genome Evolution, Life History, Molecular Evolution, Phylogenetics / Phylogenomics | Tatiana Giraud | 2017-06-28 13:40:52 | ||
05 May 2020
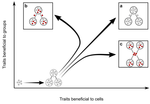
Meta-population structure and the evolutionary transition to multicellularityThe ecology of evolutionary transitions to multicellularityRecommended by Dustin Brisson based on reviews by 2 anonymous reviewersThe evolutionary transition to multicellular life from free-living, single-celled ancestors has occurred independently in multiple lineages [1-5]. This evolutionary transition to cooperative group living can be difficult to explain given the fitness advantages enjoyed by the non-cooperative, single-celled organisms that still numerically dominate life on earth [1,6,7]. Although several hypotheses have been proposed to explain the transition to multicellularity, a common theme is the abatement of the efficacy of natural selection among the single cells during the free-living stage and the promotion of the efficacy of selection among groups of cells during the cooperative stage, an argument reminiscent of those from George Williams’ seminal book [8,9]. The evolution of life cycles appears to be a key step in the transition to multicellularity as it can align fitness advantages of the single-celled 'reproductive' stage with that of the cooperative 'organismal' stage [9-12]. That is, the evolution of life cycles allows natural selection to operate over timescales longer than that of the doubling time of the free-living cells [13]. Despite the importance of this issue, identifying the range of ecological conditions that reduce the importance of natural selection at the single-celled, free-living stage and increase the importance of selection among groups of cooperating cells has not been addressed empirically. References [1] Maynard Smith, J. and Szathmáry, E. (1995). The Major Transitions in Evolution. Oxford, UK: Freeman. p 346. | Meta-population structure and the evolutionary transition to multicellularity | Caroline J Rose, Katrin Hammerschmidt, Yuriy Pichugin and Paul B Rainey | <p>The evolutionary transition to multicellularity has occurred on numerous occasions, but transitions to complex life forms are rare. While the reasons are unclear, relevant factors include the intensity of within- versus between-group selection ... | | Adaptation, Evolutionary Dynamics, Experimental Evolution | Dustin Brisson | 2019-04-04 12:26:36 | ||
18 May 2018

Modularity of genes involved in local adaptation to climate despite physical linkageDifferential effect of genes in diverse environments, their role in local adaptation and the interference between genes that are physically linkedRecommended by Sebastian Ernesto Ramos-Onsins based on reviews by Tanja Pyhäjärvi and 1 anonymous reviewerThe genome of eukaryotic species is a complex structure that experience many different interactions within itself and with the surrounding environment. The genetic architecture of a phenotype (that is, the set of genetic elements affecting a trait of the organism) plays a fundamental role in understanding the adaptation process of a species to, for example, different climate environments, or to its interaction with other species. Thus, it is fundamental to study the different aspects of the genetic architecture of the species and its relationship with its surronding environment. Aspects such as modularity (the number of genetic units and the degree to which each unit is affecting a trait of the organism), pleiotropy (the number of different effects that a genetic unit can have on an organism) or linkage (the degree of association between the different genetic units) are essential to understand the genetic architecture and to interpret the effects of selection on the genome. Indeed, the knowledge of the different aspects of the genetic architecture could clarify whether genes are affected by multiple aspects of the environment or, on the contrary, are affected by only specific aspects [1,2]. The work performed by Lotterhos et al. [3] sought to understand the genetic architecture of the adaptation to different environments in lodgepole pine (Pinus contorta), considering as candidate SNPs those previously detected as a result of its extreme association patterns to different environmental variables or to extreme population differentiation. This consideration is very important because the study is only relevant if the studied markers are under the effect of selection. Otherwise, the genetic architecture of the adaptation to different environments would be masked by other (neutral) kind of associations that would be difficult to interpret [4,5]. In order to understand the relationship between genetic architecture and adaptation, it is relevant to detect the association networks of the candidate SNPs with climate variables (a way to measure modularity) and if these SNPs (and loci) are affected by single or multiple environments (a way to measure pleiotropy). The authors used co-association networks, an innovative approach in this field, to analyse the interaction between the environmental information and the genetic polymorphism of each individual. This methodology is more appropriate than other multivariate methods - such as analysis based on principal components - because it is possible to cluster SNPs based on associations with similar environmental variables. In this sense, the co-association networks allowed to both study the genetic and physical linkage between different co-associations modules but also to compare two different models of evolution: a Modular environmental response architecture (specific genes are affected by specific aspects of the environment) or a Universal pleiotropic environmental response architecture (all genes are affected by all aspects of the environment). The representation of different correlations between allelic frequency and environmental factors (named galaxy biplots) are especially informative to understand the effect of the different clusters on specific aspects of the environment (for example, the co-association network ‘Aridity’ shows strong associations with hot/wet versus cold/dry environments). The analysis performed by Lotterhos et al. [3], although it has some unavoidable limitations (e.g., only extreme candidate SNPs are selected, limiting the results to the stronger effects; the genetic and physical map is incomplete in this species), includes relevant results and also implements new methodologies in the field. To highlight some of them: the preponderance of a Modular environmental response architecture (evolution in separated modules), the detection of physical linkage among SNPs that are co-associated with different aspects of the environment (which was unexpected a priori), the implementation of co-association networks and galaxy biplots to see the effect of modularity and pleiotropy on different aspects of environment. Finally, this work contains remarkable introductory Figures and Tables explaining unambiguously the main concepts [6] included in this study. This work can be treated as a starting point for many other future studies in the field. References [1] Hancock AM, Brachi B, Faure N, Horton MW, Jarymowycz LB, Sperone FG, Toomajian C, Roux F & Bergelson J. 2011. Adaptation to climate across the Arabidopsis thaliana genome. Science 334: 83–86. doi: 10.1126/science.1209244 | Modularity of genes involved in local adaptation to climate despite physical linkage | Katie E. Lotterhos, Sam Yeaman, Jon Degner, Sally Aitken, Kathryn Hodgins | <p>Background: Physical linkage among genes shaped by different sources of selection is a fundamental aspect of genetic architecture. Theory predicts that evolution in complex environments selects for modular genetic architectures and high recombi... | | Adaptation, Bioinformatics & Computational Biology, Genome Evolution | Sebastian Ernesto Ramos-Onsins | 2017-10-15 19:21:57 | ||
06 Jun 2019
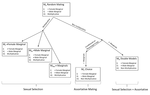
Multi-model inference of non-random mating from an information theoretic approachTell me who you mate with, I’ll tell you what’s going onRecommended by Sara Magalhaes and Alexandre Courtiol based on reviews by Alexandre Courtiol and 2 anonymous reviewersThe study of sexual selection goes as far as Darwin himself. Since then, elaborate theories concerning both intra- and inter-sexual sexual have been developed, and elegant experiments have been designed to test this body of theory. It may thus come as a surprise that the community is still debating on the correct way to measure simple components of sexual selection, such as the Bateman gradient (i.e., the covariance between the number of matings and the number of offspring)[1,2], or to quantify complex behaviours such as mate choice (the non-random choice of individuals with particular characters as mates)[3,4] and their consequences. References [1] Bateman, A. J. (1948). Intra-sexual selection in Drosophila. Heredity, 2(3), 349-368. doi: 10.1038/hdy.1948.21 | Multi-model inference of non-random mating from an information theoretic approach | Antonio Carvajal-Rodríguez | <p>Non-random mating has a significant impact on the evolution of organisms. Here, I developed a modelling framework for discrete traits (with any number of phenotypes) to explore different models connecting the non-random mating causes (mate comp... | | Evolutionary Ecology, Evolutionary Theory, Sexual Selection | Sara Magalhaes | 2019-02-08 19:24:03 |




