Replication-independent mutations: a universal signature ?
Recommended by Nicolas Galtier based on reviews by Marc Robinson-Rechavi and Robert Lanfear
Mutations are the primary source of genetic variation, and there is an obvious interest in characterizing and understanding the processes by which they appear. One particularly important question is the relative abundance, and nature, of replication-dependent and replication-independent mutations - the former arise as cells replicate due to DNA polymerization errors, whereas the latter are unrelated to the cell cycle. A recent experimental study in fission yeast identified a signature of mutations in quiescent (=non-replicating) cells: the spectrum of such mutations is characterized by an enrichment in insertions and deletions (indels) compared to point mutations, and an enrichment of deletions compared to insertions [2].
What Achaz et al. [1] report here is that the very same signature is detectable in humans. This time the approach is indirect and relies on two key aspects of mammalian reproduction biology: (1) oocytes remain quiescent over most of a female's lifespan, whereas spermatocytes keep dividing after male puberty, and (2) X chromosome, Y chromosome and autosomes spend different amounts of time in a female vs. male context. In agreement with the yeast study, Achaz et al. show that in humans the male-associated Y chromosome, for which quiescence is minimal, has by far the lowest ratios of indels to point mutations and of deletions to insertions, whereas the female-associated X chromosome has the highest. This is true both of variants that are polymorphic among humans and of fixed differences between humans and chimpanzees.
So we appear to be here learning about an important and general aspect of the mutation process. The authors suggest that, to a large extent, chromosomes tend to break in pieces at a rate that is proportional to absolute time - because indels in quiescent stage presumably result from double-strand DNA breaks. A very recent analysis of numerous mother-father-child trios in humans confirms this prediction in demonstrating an effect of maternal age, but not of paternal age, on the recombination rate [3]. This result also has important implications with respect to the interpretation of substitution rate variation among taxa and genomic compartments, particularly mitochondrial vs. nuclear, and their relationship with the generation time and longevity of organisms (e.g. [4]).
References
[1] Achaz, G., Gangloff, S., and Arcangioli, B. (2019). The quiescent X, the replicative Y and the Autosomes. BioRxiv, 351288, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/351288
[2] Gangloff, S., Achaz, G., Francesconi, S., Villain, A., Miled, S., Denis, C., and Arcangioli, B. (2017). Quiescence unveils a novel mutational force in fission yeast. eLife, 6:e27469. doi: 10.7554/eLife.27469
[3] Halldorsson, B. V., Palsson, G., Stefansson, O. A., Jonsson, H., Hardarson, M. T., Eggertsson, H. P., … Stefansson, K. (2019). Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science, 363: eaau1043. doi: 10.1126/science.aau1043
[4] Saclier, N., François, C. M., Konecny-Dupré, L., Lartillot, N., Guéguen, L., Duret, L., … Lefébure, T. (2019). Life History Traits Impact the Nuclear Rate of Substitution but Not the Mitochondrial Rate in Isopods. Molecular Biology and Evolution, in press. doi: 10.1093/molbev/msy247
| The quiescent X, the replicative Y and the Autosomes | Guillaume Achaz, Serge Gangloff, Benoit Arcangioli | <p>From the analysis of the mutation spectrum in the 2,504 sequenced human genomes from the 1000 genomes project (phase 3), we show that sexual chromosomes (X and Y) exhibit a different proportion of indel mutations than autosomes (A), ranking the... |  | Bioinformatics & Computational Biology, Genome Evolution, Human Evolution, Molecular Evolution, Population Genetics / Genomics, Reproduction and Sex | Nicolas Galtier | | 2018-07-25 10:37:48 | View |
The Red Queen lives: epistasis between linked resistance loci
Metzger CMJA, Luijckx P, Bento G, Mariadassou M, Ebert D.
10.1111/evo.12854
Evidence of epistasis provides further support to the Red Queen theory of host-parasite coevolution
Recommended by Adele Mennerat and Thierry Lefèvre
According to the Red Queen theory of antagonistic host-parasite coevolution, adaptation of parasites to the most common host genotype results in negative frequency-dependent selection whereby rare host genotypes are favoured. Assuming that host resistance relies on a genetic host-parasite (mis)match involving several linked loci, then recombination appears as much more efficient than parthenogenesis in generating new resistant host genotypes. This has long been proposed to explain one of the biggest so-called paradoxes in evolutionary biology, i.e. the maintenance of recombination despite its twofold cost.
Evidence from various study systems indicates that successful infection (and hence host resistance) depends on a genetic match between the parasite’s and the host’s genotype via molecular interactions involving elicitor/receptor mechanisms. However the key assumption of epistasis, i.e. that this genetic host-parasite match involves several linked resistance loci, remained unsupported so far. Metzger and coauthors [1] now provide empirical support for it.
Daphnia magna can reproduce both sexually and clonally and their well-studied interaction with Pasteuria ramosa makes them an excellent model system to investigate the genetics of host resistance. D. magna hosts were found to be either resistant (complete lack of attachment of parasite spores to the host’s foregut) or susceptible (full attachment). In this study the authors carried out an elegant Mendelian genetic investigation by performing multiple crosses between four host genotypes differing in their resistance to two different parasite isolates [1].
Their results show that resistance of D. magna to each of the two P. ramosa isolates relies on Mendelian inheritance at two loci that are linked (A and B), each of them having two alleles with dominant resistance; furthermore resistance to one parasite isolate confers susceptibility to the other. They also show that a third locus appears to confer double resistance (C), but that even double resistant hosts remain susceptible to other parasite isolates, and hence that universal host resistance is lacking – all of this supporting the Red Queen theory.
This paper demonstrates with a high level of clarity that host resistance is governed by multiple linked loci. The assumption of epistasis between resistance loci is supported, which makes it possible for sexual recombination to be maintained by antagonistic host-parasite coevolution.
Reference
[1] Metzger CMJA, Luijckx P, Bento G, Mariadassou M, Ebert D. 2016. The Red Queen lives: epistasis between linked resistance loci. Evolution 70:480-487. doi: 10.1111/evo.12854
| The Red Queen lives: epistasis between linked resistance loci | Metzger CMJA, Luijckx P, Bento G, Mariadassou M, Ebert D. | A popular theory explaining the maintenance of genetic recombination (sex) is the Red Queen Theory. This theory revolves around the idea that time-lagged negative frequency-dependent selection by parasites favors rare host genotypes generated thro... |  | Evolutionary Dynamics, Evolutionary Theory, Reproduction and Sex, Species interactions | Adele Mennerat | | 2016-12-14 13:58:53 | View |
Thermal regimes, but not mean temperatures, drive patterns of rapid climate adaptation at a continent-scale: evidence from the introduced European earwig across North America
Jean-Claude Tourneur, Joël Meunier
https://doi.org/10.1101/550319
Temperature variance, rather than mean, drives adaptation to local climate
Recommended by Fabien Aubret based on reviews by Ben Phillips and Eric Gangloff
Climate change is impacting eco-systems worldwide and driving many populations to move, adapt or go extinct. It is increasingly appreciated, for example, that species may adjust their phenology in response to climate change, although empirical data is scarce. In this preprint [1], Tourneur and Meunier report an impressive sampling effort in which life-history traits were measured across introduced populations of earwig in North America. The authors examine whether variation in life-history across populations is correlated with aspects of the thermal climate experienced by each population: mean temperature and seasonality of temperature. They find some fascinating correlations between life-history and thermal climate; correlations with the seasonality of temperature, but not with mean temperature. This study provides relatively uncommon data, in the sense that where most of the literature looking at adaptation in animals in response to climate change has focused on physiological traits [2, 3], this study examines changes in life-history traits with time scales relevant to impending climate change, and provides a reasonable argument that this is adaptation, not just constraint.
References
[1] Tourneur, J.-C. and Meunier, J. (2019). Thermal regimes, but not mean temperatures, drive patterns of rapid climate adaptation at a continent-scale: evidence from the introduced European earwig across North America. BioRxiv, 550319, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/550319
[2] Kellermann, V., Overgaard, J., Hoffmann, A. A., Fløjgaard, C., Svenning, J. C., & Loeschcke, V. (2012). Upper thermal limits of Drosophila are linked to species distributions and strongly constrained phylogenetically. Proceedings of the National Academy of Sciences, 109(40), 16228-16233. doi: 10.1073/pnas.1207553109
[3] Hoffmann, A. A., & Sgro, C. M. (2011). Climate change and evolutionary adaptation. Nature, 470(7335), 479. doi: 10.1038/nature09670
| Thermal regimes, but not mean temperatures, drive patterns of rapid climate adaptation at a continent-scale: evidence from the introduced European earwig across North America | Jean-Claude Tourneur, Joël Meunier | <p>The recent development of human societies has led to major, rapid and often inexorable changes in the environment of most animal species. Over the last decades, a growing number of studies formulated predictions on the modalities of animal adap... |  | Adaptation, Evolutionary Ecology, Life History | Fabien Aubret | | 2019-02-15 09:12:11 | View |
Y chromosome makes fruit flies die younger
Recommended by Gabriel Marais, Jean-François Lemaitre and Cristina Vieira
In most animal species, males and females display distinct survival prospect, a phenomenon known as sex gap in longevity (SGL, Marais et al. 2018). The study of SGLs is crucial not only for having a full picture of the causes underlying organisms’ health, aging and death but also to initiate the development of sex-specific anti-aging interventions in humans (Austad and Bartke 2015). Three non-mutually evolutionary causes have been proposed to underlie SGLs (Marais et al. 2018). First, SGLs could be the consequences of sex-differences in life history strategies. For example, evolving dimorphic traits (e.g. body size, ornaments or armaments) may imply unequal physiological costs (e.g. developmental, maintenance) between the sexes and this may result in differences in longevity and aging. Second, mitochondria are usually transmitted by the mother and thus selection is blind to mitochondrial deleterious mutations affecting only males. Such mutations can freely accumulate in the mitochondrial genome and may reduce male longevity, a phenomenon called the mother’s curse (Frank and Hurst 1996). Third, in species with sex chromosomes, all recessive deleterious mutations will be expressed on the single X chromosome in XY males and may reduce their longevity (the unguarded X effect). In addition, the numerous transposable elements (TEs) on the Y chromosome may affect aging. TE activity is normally repressed by epigenetic regulation (DNA methylation, histone modifications and small RNAs). However, it is known that this regulation is disrupted with increasing age. Because of the TE-rich Y chromosome, more TEs may become active in old males than in old females, generating more somatic mutations, accelerating aging and reducing longevity in males (the toxic Y effect, Marais et al. 2018).
The relative contributions of these different effects to SGLs remain unknown. Sex-differences in life history strategies have been considered as the most important cause of SGLs for long (Tidière et al. 2015) but this effect remain equivocal (Lemaître et al. 2020) and cannot explain alone the diversity of patterns observed across species (Marais et al. 2018). Similarly, while studies in Drosophila and humans have shown that the mother’s curse contributes to SGLs in those organisms (e.g. Milot et al. 2007), its contribution may not be strong. Recently, two large-scale comparative analyses have shown that in species with XY chromosomes males show a shorter lifespan compared to females, while in species with ZW chromosomes (a system in which the female are the heterogametic sex and are ZW, and the males ZZ) the opposite pattern is observed (Pipoly et al. 2015; Xirocostas et al. 2020). Apart from these correlational studies, very little experimental tests of the effect of sex chromosomes on longevity have been conducted. In Drosophila, the evidence suggests that the unguarded X effect does not contribute to SGLs (Brengdahl et al. 2018). Whether a toxic Y effect exists in this species was unknown.
In a very elegant study, Brown et al. (2020) provided strong evidence for such a toxic Y effect in Drosophila melanogaster. First, they checked that in the D. melanogaster strain that they were studying (Canton-S), males were indeed dying younger than females. They also confirmed that in this strain, as in others, the male genomes include more repeats and heterochromatin than the female ones using cytometry. A careful analysis of the heterochromatin (using H3K9me2, a repressive histone modification typical of heterochromatin, as a proxy) in old flies revealed that heterochromatin loss was much more important in males than in females, in particular on the Y chromosome (but also to a lesser extent at the pericentromric regions of the autosomes). This change in heterochromatin had two outputs, they found. First, the expression of the genes in those regions was affected. They highlighted that many of such genes are involved in immunity and regulation with a potential impact on longevity. Second, they found a striking TE reactivation. These two effects were stronger in males. While females showed clear reactivation of 6 TEs, with the total fraction of repeats in the transcriptome going from 2% (young females) to 4.6% (old females), males experienced the reactivation of 32 TEs, with the total fraction of repeats in the transcriptome going from 1.6% (young males) to 5.8% (old males). It appeared that most of these TEs are Y-linked. And when focusing on Y-linked repeats, they found that 32 Y-linked TEs became upregulated during male aging and the fraction of Y-linked TEs in the transcriptome increased ninefold.
All these observations clearly suggested that male longevity was decreased because of a toxic Y effect. To really uncover a causal relationship between having a Y chromosome and shorter longevity, Brown et al. (2020) artificially produced flies with atypical karyotypes: X0 males, XXY females and XYY males. This is very interesting as they could uncouple the effect of the phenotypical sex (being male or female) and having a Y chromosome or not, as in fruit flies sex is determined not by the Y chromosome but by the X/autosome ratio. Their results are striking. They found that longevity of the X0 males was the highest (higher than XX females in fact), and that of the XYY males the lowest. Females XXY had intermediate longevities. Importantly, this was found to be robust to genomic background as results were the same using crosses from different strains. When analysing TEs of these flies, they found a particularly strong expression of the Y-linked TEs in old XXY and XYY flies. Interestingly, in young XXY and XYY flies Y-linked TEs expression was also strong, suggesting the chromatin regulation of the Y chromosome is disrupted in these flies.
This work points to the idea that SGLs in D. melanogaster are mainly explained by the toxic Y effect. The molecular details however remain to be elucidated. The effect of the Y chromosome on aging might be more complex than envisioned in the toxic Y model presented above. Brown et al. (2020) indeed found that heterochromatin loss was globally faster in males, both at the Y chromosome and the autosomes. The organisation of the nucleus, in particular of the nucleolus, which is involved in heterochromatin maintenance, involves the sex chromosomes in D. melanogaster as discussed in the paper, and may explain this observation. The epigenetic status of the Y chromosome is known to affect that of all the autosomes in Drosophila (Lemos et al. 2008). Also, in Brown et al. (2020) most of the work (in particular the genomic part) has been done on Canton-S. Only D. melanogaster was studied but limited data suggest different Drosophila species may have different SGLs. The TE analysis is known to be tricky, different tools to analyse TE expression exist (e.g. Lerat et al. 2017; Lanciano and Cristofari 2020). Future work should focus on testing the toxic Y effect on other D. melanogaster strains and other Drosophila species, using different tools to study TE expression, and on dissecting the molecular details of the toxic Y effect.
References
Austad, S. N., and Bartke, A. (2015). Sex differences in longevity and in responses to anti-aging interventions: A Mini-Review. Gerontology, 62(1), 40–46. 10.1159/000381472
Brengdahl, M., Kimber, C. M., Maguire-Baxter, J., and Friberg, U. (2018). Sex differences in life span: Females homozygous for the X chromosome do not suffer the shorter life span predicted by the unguarded X hypothesis. Evolution; international journal of organic evolution, 72(3), 568–577. 10.1111/evo.13434
Brown, E. J., Nguyen, A. H., and Bachtrog, D. (2020). The Y chromosome may contribute to sex-specific ageing in Drosophila. Nature ecology and evolution, 4(6), 853–862. 10.1038/s41559-020-1179-5 or preprint link on bioRxiv
Frank, S. A., and Hurst, L. D. (1996). Mitochondria and male disease. Nature, 383(6597), 224. 10.1038/383224a0
Lanciano, S., and Cristofari, G. (2020). Measuring and interpreting transposable element expression. Nature reviews. Genetics, 10.1038/s41576-020-0251-y. Advance online publication. 10.1038/s41576-020-0251-y
Lemaître, J. F., Ronget, V., Tidière, M., Allainé, D., Berger, V., Cohas, A., Colchero, F., Conde, D. A., Garratt, M., Liker, A., Marais, G., Scheuerlein, A., Székely, T., and Gaillard, J. M. (2020). Sex differences in adult lifespan and aging rates of mortality across wild mammals. Proceedings of the National Academy of Sciences of the United States of America, 117(15), 8546–8553. 10.1073/pnas.1911999117
Lemos, B., Araripe, L. O., and Hartl, D. L. (2008). Polymorphic Y chromosomes harbor cryptic variation with manifold functional consequences. Science (New York, N.Y.), 319(5859), 91–93. 10.1126/science.1148861
Lerat, E., Fablet, M., Modolo, L., Lopez-Maestre, H., and Vieira, C. (2017). TEtools facilitates big data expression analysis of transposable elements and reveals an antagonism between their activity and that of piRNA genes. Nucleic acids research, 45(4), e17. 10.1093/nar/gkw953
Marais, G., Gaillard, J. M., Vieira, C., Plotton, I., Sanlaville, D., Gueyffier, F., and Lemaitre, J. F. (2018). Sex gap in aging and longevity: can sex chromosomes play a role?. Biology of sex differences, 9(1), 33. 10.1186/s13293-018-0181-y
Milot, E., Moreau, C., Gagnon, A., Cohen, A. A., Brais, B., and Labuda, D. (2017). Mother's curse neutralizes natural selection against a human genetic disease over three centuries. Nature ecology and evolution, 1(9), 1400–1406. 10.1038/s41559-017-0276-6
Pipoly, I., Bókony, V., Kirkpatrick, M., Donald, P. F., Székely, T., and Liker, A. (2015). The genetic sex-determination system predicts adult sex ratios in tetrapods. Nature, 527(7576), 91–94. 10.1038/nature15380
Tidière, M., Gaillard, J. M., Müller, D. W., Lackey, L. B., Gimenez, O., Clauss, M., and Lemaître, J. F. (2015). Does sexual selection shape sex differences in longevity and senescence patterns across vertebrates? A review and new insights from captive ruminants. Evolution; international journal of organic evolution, 69(12), 3123–3140. 10.1111/evo.12801
Xirocostas, Z. A., Everingham, S. E., and Moles, A. T. (2020). The sex with the reduced sex chromosome dies earlier: a comparison across the tree of life. Biology letters, 16(3), 20190867. 10.1098/rsbl.2019.0867
| The Y chromosome may contribute to sex-specific ageing in Drosophila | Emily J Brown, Alison H Nguyen, Doris Bachtrog | <p>Heterochromatin suppresses repetitive DNA, and a loss of heterochromatin has been observed in aged cells of several species, including humans and *Drosophila*. Males often contain substantially more heterochromatic DNA than females, due to the ... |  | Bioinformatics & Computational Biology, Expression Studies, Genetic conflicts, Genome Evolution, Genotype-Phenotype, Molecular Evolution, Reproduction and Sex | Gabriel Marais | | 2020-07-28 15:06:18 | View |
Things softly attained are long retained: Dissecting the Impacts of Selection Regimes on Polymorphism Maintenance in Experimental Spatially Heterogeneous Environments
Romain Gallet, Rémy Froissart, Virginie Ravigné
https://doi.org/10.1101/100743
Experimental test of the conditions of maintenance of polymorphism under hard and soft selection
Recommended by Stephanie Bedhomme based on reviews by Joachim Hermisson and 2 anonymous reviewers
Theoretical work, initiated by Levene (1953) [1] and Dempster (1955) [2], suggests that within a given environment, the way populations are regulated and contribute to the next generation is a key factor for the maintenance of local adaptation polymorphism. In this theoretical context, hard selection describes the situation where the genetic composition of each population affects its contribution to the next generation whereas soft selection describes the case where the contribution of each population is fixed, whatever its genetic composition. Soft selection is able to maintain polymorphism, whereas hard selection invariably leads to the fixation of one of the alleles. Although the specific conditions (e.g. of migration between populations or drift level) in which this prediction holds have been studied in details by theoreticians, experimental tests have mainly failed, usually leading to the conclusion that the allele frequency dynamics was driven by other mechanisms in the experimental systems and conditions used.
Gallet, Froissart and Ravigné [3] have set up a bacterial experimental system which allowed them to convincingly demonstrate that soft selection generates the conditions for polymorphism maintenance when hard selection does not, everything else being equal. The key ingredients of their experimental system are (1) the possibility to accurately produce hard and soft selection regimes when daily transferring the populations and (2) the ability to establish artificial well-characterized reproducible trade-offs. To do so, they used two genotypes resisting each one to one antibiotic and combined, across habitats, low antibiotic doses and difference in medium productivity. The experimental approach contains two complementary parts: the first one is looking at changes in the frequencies of two genotypes, initially introduced at around 50% each, over a small number of generations (ca 40) in different environments and selection regimes (soft/hard) and the second one is convincingly showing polymorphism protection by establishing that in soft selection regimes, the lowest fitness genotype is not eliminated even when introduced at low frequency.
In this manuscript, a key point is the dialog between theoretical and experimental approaches. The experiments have been thought and designed to be as close as possible to the situations analysed in theoretical work. For example, the experimental polymorphism protection test (experiment 2) closely matches the equivalent analysis classically performed in theoretical approaches. This close fit between theory and experiment is clearly a strength of this study. This said, the experimental system allowing them to realise this close match also has some limitations. For example, changes in allele frequencies could only be monitored over a quite low number of generations because a longer time-scale would have allowed the contribution of de novo mutations and the likely emergence of a generalist genotype resisting to both antibiotics used to generate the local adaptation trade-offs. These limitations, as well as the actual significance of the experimental tests, are discussed in deep details in the manuscript.
References
[1] Levene H. 1953. Genetic equilibrium when more than one niche is available. American Naturalist 87: 331–333. doi: 10.1086/281792
[2] Dempster ER. 1955. Maintenance of genetic heterogeneity. Cold Spring Harbor Symposia on Quantitative Biology. 20: 25–32. doi: 10.1101/SQB.1955.020.01.005
[3] Gallet R, Froissart R, Ravigné V. 2017. Things softly attained are long retained: dissecting the impacts of selection regimes on polymorphism maintenance in experimental spatially heterogeneous environments. bioRxiv 100743; doi: 10.1101/100743
| Things softly attained are long retained: Dissecting the Impacts of Selection Regimes on Polymorphism Maintenance in Experimental Spatially Heterogeneous Environments | Romain Gallet, Rémy Froissart, Virginie Ravigné | <p>Predicting and managing contemporary adaption requires a proper understanding of the determinants of genetic variation. Spatial heterogeneity of the environment may stably maintain polymorphism when habitat contribution to the next generation c... | 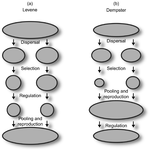 | Adaptation, Evolutionary Theory | Stephanie Bedhomme | | 2017-01-17 11:06:21 | View |
Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between
Fanny Pouyet and Kimberly J. Gilbert
http://arxiv.org/abs/1909.11490
Molecular evolution through the joint lens of genomic and population processes.
Recommended by Guillaume Achaz based on reviews by Benoit Nabholz and 1 anonymous reviewer
In their perspective article, F Pouyet and KJ Gilbert (2020), propose an interesting overview of all the processes that sculpt patterns of molecular evolution. This well documented article covers most (if not all) important facets of the recurrent debate that has marked the history of molecular evolution: the relative importance of natural selection and neutral processes (i.e. genetic drift). I particularly enjoyed reading this review, that instead of taking a clear position on the debate, catalogs patiently every pieces of information that can help understand how patterns we observed at the genome level, can be understood from a selectionnist point of view, from a neutralist one, and, to quote their title, from "everything in between". The review covers the classical objects of interest in population genetics (genetic drift, selection, demography and structure) but also describes several genomic processes (meiotic drive, linked selection, gene conversion and mutation processes) that obscure the interpretation of these population processes. The interplay between all these processes is very complex (to say the least) and have resulted in many cases in profound confusions while analyzing data. It is always very hard to fully acknowledge our ignorance and we have many times payed the price of model misspecifications. This review has the grand merit to improve our awareness in many directions. Being able to cover so many aspects of a wide topic, while expressing them simply and clearly, connecting concepts and observations from distant fields, is an amazing "tour de force". I believe this article constitutes an excellent up-to-date introduction to the questions and problems at stake in the field of molecular evolution and will certainly also help established researchers by providing them a stimulating overview supported with many relevant references.
References
[1] Pouyet F, Gilbert KJ (2020) Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between. arXiv:1909.11490 [q-bio]. ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. url:https://arxiv.org/abs/1909.11490
| Towards an improved understanding of molecular evolution: the relative roles of selection, drift, and everything in between | Fanny Pouyet and Kimberly J. Gilbert | <p>A major goal of molecular evolutionary biology is to identify loci or regions of the genome under selection versus those evolving in a neutral manner. Correct identification allows accurate inference of the evolutionary process and thus compreh... |  | Genome Evolution, Population Genetics / Genomics | Guillaume Achaz | | 2019-09-26 10:58:10 | View |
Another step towards grasping the complexity of the environmental response of traits
Recommended by Benoit Pujol based on reviews by 2 anonymous reviewers
One can only hope that one day, we will be able to evaluate how the ecological complexity surrounding natural populations affects their ability to adapt. This is more like a long term quest than a simple scientific aim. Many steps are heading in the right direction. This paper by Monroe and colleagues (2021) is one of them.
Many ecological and genetic mechanisms shape the evolutionary potential of phenotypic trait variation and many of them involve environmental heterogeneity (Pujol et al 2018). To date, we cannot look into these ecological and genetic mechanisms without oversimplifying their effects. We often look into trait variation one trait at a time albeit the variation of multiple phenotypic traits is often linked at the genetic or environmental level. As a consequence, we put our conclusions at risk by not accounting for the reciprocal impacts of trait changes upon each other (Teplitsky et al 2014). We also usually restrict the study of a continuous gradient of environmental conditions to a few conditions because it would otherwise be impossible to model its environmental effect. As a consequence, we miss the full picture of the continuous often nonlinear phenotypic plastic response. Whether the trait undergo threshold effect changes thereby remains obscured to us. Collectively, these issues impede our ability to understand how selection shapes the ecological strategy of organisms under variable environments.
In this paper, Monroe and colleagues (2021) propose an original approach that raised to these two challenges. They analysed phenotypic plastic changes in response to a continuous environment in a multidimensional trait space, namely the response of Brachypodium plant developmental and physiological traits to a continuous gradient of soil moisture. They used dry down experimental treatments to produce the continuous soil moisture gradient and compared the plant capacity to use water between annual B. distachyon and perennial B. sylvaticum. Their results revealed the best mathematical functions that model the nonlinear curvature of the continuous plastic response of Brachypodium plants. This work reinforces our view that nonlinear plastic responses can result in greater or lesser trait values at any stage of the environmental gradient that were unexpected on the basis of linear predictors (Gienapp and Brommer 2014). Their findings also imply that different threshold responses characterize different genotypes. These could otherwise have been missed by a classical approach. By shedding light on unforeseen interactions between traits that make their correlation vary along the nonlinear response, they were able to describe more accurately Brachypodium ecological strategies and the changes in evolutionary constraints along the soil moisture gradient.
Their empirical approach allows to test what environmental conditions maximises the opportunity for selection to shape trait variation. For example, it revealed unforeseen divergence in potentially adaptive mechanisms or life history strategies – and not just trait values – between annual and perennial species of Brachypodium. Behind every environmental variation of the constraints to the future evolutionary change of multiple traits, we can expect that the evolutionary history of the populations shaped their trait genetic correlations. Investigating the nonlinear signature of adaptive evolution across continuous environments will get us into uncharted territory.
Our ability to predict the adaptive potential of species is limited. With their approach of continuous environmental gradients beyond linearity, Monroe and collaborators (2021) improve our understanding of plant phenotypic responses and open a brand new range of exciting developments. As they mention: "the opportunity for scaling up" their approach is big. To illustrate this prospect, I can easily think of an example: the quantitative genetic random regression model. This model allows to use any degree of genetic relatedness in a wild population to estimate the genetic variation of phenotypic plastic reaction norms (Nussey et al 2007, Pujol and Galaud 2013). However, in this approach, only a few modalities of the environmental gradient are used to model nonlinear phenotypic plastic responses. From there, it is rather intuitive. Combining the best of these two approaches (continuity of genetic relatedness in the wild & continuity of environmental gradient in experiments) could open ground breaking new perspectives in research.
References
Gienapp P. & J.E. Brommer. 2014. Evolutionary dynamics in response to climate change. In: Charmentier A, Garant D, Kruuk LEB, editors. Quantitative genetics in the wild. Oxford: Oxford University Press, Oxford. pp. 254–273. doi: https://doi.org/10.1093/acprof:oso/9780199674237.003.0015
Monroe, J. G., Cai, H., and Des Marais, D. L. (2020). Trait plasticity and covariance along a continuous soil moisture gradient. bioRxiv, 2020.02.17.952853, ver. 5 peer-reviewed and recommended by PCI Evol Biol. doi: https://doi.org/10.1101/2020.02.17.952853
Pujol et al. (2018). The missing response to selection in the wild. Trends in ecology & evolution, 33(5), 337-346. doi: https://doi.org/10.1016/j.tree.2018.02.007
Pujol, B., and Galaud, J. P. (2013). A practical guide to quantifying the effect of genes underlying adaptation in a mixed genomics and evolutionary ecology approach. Botany Letters, 160(3-4), 197-204. doi: https://doi.org/10.1080/12538078.2013.799045
Nussey, D. H., Wilson, A. J., and Brommer, J. E. (2007). The evolutionary ecology of individual phenotypic plasticity in wild populations. Journal of evolutionary biology, 20(3), 831-844. doi: https://doi.org/10.1111/j.1420-9101.2007.01300.x
Teplitsky et al. (2014). Assessing multivariate constraints to evolution across ten long-term avian studies. PLoS One, 9(3), e90444. doi: https://doi.org/10.1371/journal.pone.0090444
| Trait plasticity and covariance along a continuous soil moisture gradient | J Grey Monroe, Haoran Cai, David L Des Marais | <p>Water availability is perhaps the greatest environmental determinant of plant yield and fitness. However, our understanding of plant-water relations is limited because it is primarily informed by experiments considering soil moisture variabilit... |  | Phenotypic Plasticity | Benoit Pujol | | 2020-02-20 16:34:40 | View |
Trade-offs in fitness components and ecological source-sink dynamics affect host specialisation in two parasites of Artemia shrimps
Recommended by Frédéric Guillaume based on reviews by Anne Duplouy, Seth Barribeau and Cindy Gidoin based on reviews by Anne Duplouy, Seth Barribeau and Cindy Gidoin
Ecological specialisation, especially among parasites infecting a set of host species, is ubiquitous in nature. Host specialisation can be understood as resulting from trade-offs in parasite infectivity, virulence and growth. However, it is not well understood how variation in these trade-offs shapes the overall fitness trade-off a parasite faces when adapting to multiple hosts. For instance, it is not clear whether a strong trade-off in one fitness component may sufficiently constrain the evolution of a generalist parasite despite weak trade-offs in other components. A second mechanism explaining variation in specialisation among species is habitat availability and quality. Rare habitats or habitats that act as ecological sinks will not allow a species to persist and adapt, preventing a generalist phenotype to evolve. Understanding the prevalence of those mechanisms in natural systems is crucial to understand the emergence and maintenance of host specialisation, and biodiversity in general.
In their study "Trait-specific trade-offs prevent niche expansion in two parasites", Lievens et al. [1] report the results of an evolution experiment involving two parasitic microsporidians, Anostracospora rigaudi and Enterocytospora artemiae, infecting two sympatric species of brine shrimp, Artemia franciscana and Artemia parthenogenetica. The two parasites were originally specialised on their primary host: A. rigaudi on A. parthenogenetica and E. artemiae on A. franciscana, although they encounter both species in the wild but at different rates. After passaging each parasite on each single host and on both hosts alternatively, Lievens et al. asked how host specialisation evolved. They found no change in specialisation at the fitness level in A. rigaudi in either treatment, while E. artemiae became more of a generalist after having been exposed to its secondary host, A. parthenogenetica. The most interesting part of the study is the decomposition of the fitness trade-off into its underlying trade-offs in spore production, infectivity and virulence. Both species remained specialised for spore production on their primary host, interpreted as caused by a strong trade-off between hosts preventing improvements on the secondary host. A. rigaudi evolved reduced virulence on its primary host without changes in the overall fitness trad-off, while E. artemiae evolved higher infectivity on its secondary host making it a more generalist parasite and revealing a weak trade-off for this trait and for fitness. Nevertheless, both parasites retained higher fitness on their primary host because of the lack of an evolutionary response in spore production.
This study made two important points. First, it showed that despite apparent strong trade-off in spore production, a weak trade-off in infectivity allowed E. artemiae to become less specialised. In contrast, A. rigaudi remained specialised, presumably because the strong trade-off in spore production was the overriding factor. The fitness trade-off that results from the superposition of multiple underlying trade-offs is thus difficult to predict, yet crucial to understand potential evolutionary outcomes. A second insight is related to the ecological context of the evolution of specialisation. The results showed that E. artemiae should be less specialised than observed, which points to a role played by source-sink dynamics on A. parthenogenetica in the wild. The experimental approach of Lievens et al. thus allowed them to nicely disentangle the various sources of constraints on the evolution of host adaptation in the Artemia system.
References
[1] Lievens, E.J.P., Michalakis, Y. and Lenormand, T. (2019). Trait-specific trade-offs prevent niche expansion in two parasites. bioRxiv, 621581, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/621581
| Trait-specific trade-offs prevent niche expansion in two parasites | Eva JP Lievens, Yannis Michalakis, Thomas Lenormand | <p>The evolution of host specialization has been studied intensively, yet it is still often difficult to determine why parasites do not evolve broader niches – in particular when the available hosts are closely related and ecologically similar. He... | 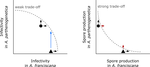 | Adaptation, Evolutionary Ecology, Evolutionary Epidemiology, Experimental Evolution, Life History, Species interactions | Frédéric Guillaume | | 2019-05-13 13:44:34 | View |
Transcriptional differences between the two host strains of Spodoptera frugiperda (Lepidoptera: Noctuidae)
Marion Orsucci, Yves Moné, Philippe Audiot, Sylvie Gimenez, Sandra Nhim, Rima Naït-Saïdi, Marie Frayssinet, Guillaume Dumont, Jean-Paul Boudon, Marin Vabre, Stéphanie Rialle, Rachid Koual, Gael J. Kergoat, Rodney N. Nagoshi, Robert L. Meagher, Emmanuelle d'Alencon, Nicolas Nègre
https://doi.org/10.1101/263186
Speciation through selection on mitochondrial genes?
Recommended by Astrid Groot based on reviews by Heiko Vogel and Sabine Haenniger
Whether speciation through ecological specialization occurs has been a thriving research area ever since Mayr (1942) stated this to play a central role. In herbivorous insects, ecological specialization is most likely to happen through host plant differentiation (Funk et al. 2002). Therefore, after Dorothy Pashley had identified two host strains in the Fall armyworm (FAW), Spodoptera frugiperda, in 1988 (Pashley 1988), researchers have been trying to decipher the evolutionary history of these strains, as this seems to be a model species in which speciation is currently occurring through host plant specialization. Even though FAW is a generalist, feeding on many different host plant species (Pogue 2002) and a devastating pest in many crops, Pashley identified a so-called corn strain and a so-called rice strain in Puerto Rico. Genetically, these strains were found to differ mostly in an esterase, although later studies showed additional genetic differences and markers, mostly in the mitochondrial COI and the nuclear TPI. Recent genomic studies showed that the two strains are overall so genetically different (2% of their genome being different) that these two strains could better be called different species (Kergoat et al. 2012). So far, the most consistent differences between the strains have been their timing of mating activities at night (Schoefl et al. 2009, 2011; Haenniger et al. 2019) and hybrid incompatibilities (Dumas et al. 2015; Kost et al. 2016). Whether and to what extent host plant preference or performance contributed to the differentiation of these sympatrically occurring strains has remained unclear.
In the current study, Orsucci et al. (2020) performed oviposition assays and reciprocal transplant experiments with both strains to measure fitness effects, in combination with a comprehensive RNAseq experiment, in which not only lab reared larvae were analysed, but also field-collected larvae. When testing preference and performance on the two host plants corn and rice, the authors did not find consistent fitness differences between the two strains, with both strains performing less on rice plants, although larvae from the corn strain survived more on corn plants than those from the rice strain. These results mostly confirm findings of a number of investigations over the past 30 years, where no consistent differences on the two host plants were observed (reviewed in Groot et al. 2016). However, the RNAseq experiments did show some striking differences between the two strains, especially in the reciprocally transplanted larvae, where both strains had been reared on rice or on corn plants for one generation: both strains showed transcriptional responses that correspond to their respective putative host plants, i.e. overexpression of genes involved in digestion and metabolic activity, and underexpression of genes involved in detoxification, in the corn strain on corn and in the rice strain on rice. Interestingly, similar sets of genes were found to be overexpressed in the field-collected larvae with which a RNAseq experiment was conducted as well.
The most interesting result of the study performed by Orsucci et al. (2020) is the underexpression in the corn strain of so-called numts, small genomic sequences that corresponded to fragments of the mitochondrial COI and COIII. These two numts were differentially expressed in the two strains in all RNAseq experiments analysed. This result coincides with the fact that the COI is one of the main diagnostic markers to distinguish these two strains. The authors suggestion that a difference in energy production between these two strains may be linked to a shift in host plant preference matches their finding that rice plants seem to be less suitable host plants than corn plants. However, as the lower suitability of rice plants was true for both strains, it remains unclear whether and how this difference could be linked to possible host plant differentiation between the strains. The authors also suggest that COI and potentially other mitochondrial genes may be the original target of selection between these two strains. This is especially interesting in light of the fact that field-collected larvae have frequently been found to have a rice strain mitochondrial genotype and a corn strain nuclear genotype, also in this study, while in the lab (female rice strain x male corn strain) hybrid females (i.e. females with a rice strain mitochondrial genotype and a corn strain nuclear genotype) are behaviorally sterile (Kost et al. 2016). Whether and how selection on mitochondrial genes or on mitonuclear interactions has indeed affected the evolution of these strains in the New world, and will affect the evolution of FAW in newly invaded habitats in the Old world, including Asia and Australia – where, so far, only corn strain and (female rice strain x male corn strain) hybrids have been found (Nagoshi 2019), will be a challenging research question for the coming years.
References
[1] Dumas, P. et al. (2015). Spodoptera frugiperda (Lepidoptera: Noctuidae) host-plant variants: two host strains or two distinct species?. Genetica, 143(3), 305-316. doi: 10.1007/s10709-015-9829-2
[2] Funk, D. J., Filchak, K. E. and Feder J. L. (2002) Herbivorous insects: model systems for the comparative study of speciation ecology. In: Etges W.J., Noor M.A.F. (eds) Genetics of Mate Choice: From Sexual Selection to Sexual Isolation. Contemporary Issues in Genetics and Evolution, vol 9. Springer, Dordrecht. doi: 10.1007/978-94-010-0265-3_10
[3] Groot, A. T., Unbehend, M., Hänniger, S., Juárez, M. L., Kost, S. and Heckel D. G.(2016) Evolution of reproductive isolation of Spodoptera frugiperda. In Allison, J. and Cardé, R. (eds) Sexual communication in moths. Chapter 20: 291-300.
[4] Hänniger, S. et al. (2017). Genetic basis of allochronic differentiation in the fall armyworm. BMC evolutionary biology, 17(1), 68. doi: 10.1186/s12862-017-0911-5
[5] Kost, S., Heckel, D. G., Yoshido, A., Marec, F., and Groot, A. T. (2016). AZ‐linked sterility locus causes sexual abstinence in hybrid females and facilitates speciation in Spodoptera frugiperda. Evolution, 70(6), 1418-1427. doi: 10.1111/evo.12940
[6] Mayr, E. (1942) Systematics and the origin of species. Columbia University Press, New York.
[7] Nagoshi, R. N. (2019). Evidence that a major subpopulation of fall armyworm found in the Western Hemisphere is rare or absent in Africa, which may limit the range of crops at risk of infestation. PloS one, 14(4). doi: 10.1371/journal.pone.0208966
[8] Orsucci, M., Moné, Y., Audiot, P., Gimenez, S., Nhim, S., Naït-Saïdi, R., Frayssinet, M., Dumont, G., Boudon, J.-P., Vabre, M., Rialle, S., Koual, R., Kergoat, G. J., Nagoshi, R. N., Meagher, R. L., d’Alençon, E. and Nègre N. (2020) Transcriptional differences between the two host strains of Spodoptera frugiperda (Lepidoptera: Noctuidae). bioRxiv, 263186, ver. 2 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/263186
[9] Pashley, D. P. (1988) Current Status of Fall Armyworm Host Strains. Florida Entomologist 71 (3): 227–34. doi: 10.2307/3495425
[10] Pogue, M. (2002). A World Revision of the Genus Spodoptera Guenée (Lepidoptera: Noctuidae). American Entomological Society.
[11] Schöfl, G., Heckel, D. G., and Groot, A. T. (2009). Time‐shifted reproductive behaviours among fall armyworm (Noctuidae: Spodoptera frugiperda) host strains: evidence for differing modes of inheritance. Journal of Evolutionary Biology, 22(7), 1447-1459. doi: 10.1111/j.1420-9101.2009.01759.x
[12] Schöfl, G., Dill, A., Heckel, D. G., and Groot, A. T. (2011). Allochronic separation versus mate choice: nonrandom patterns of mating between fall armyworm host strains. The American Naturalist, 177(4), 470-485. doi: 10.1086/658904
| Transcriptional differences between the two host strains of Spodoptera frugiperda (Lepidoptera: Noctuidae) | Marion Orsucci, Yves Moné, Philippe Audiot, Sylvie Gimenez, Sandra Nhim, Rima Naït-Saïdi, Marie Frayssinet, Guillaume Dumont, Jean-Paul Boudon, Marin Vabre, Stéphanie Rialle, Rachid Koual, Gael J. Kergoat, Rodney N. Nagoshi, Robert L. Meagher, Emm... | <p>Spodoptera frugiperda, the fall armyworm (FAW), is an important agricultural pest in the Americas and an emerging pest in sub-Saharan Africa, India, East-Asia and Australia, causing damage to major crops such as corn, sorghum and soybean. While... |  | Adaptation, Evolutionary Ecology, Expression Studies, Life History, Speciation | Astrid Groot | | 2018-05-09 13:04:34 | View |
Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network
Maud Irène Tenaillon, Khawla Sedikki, Maeva Mollion, Martine Le Guilloux, Elodie Marchadier, Adrienne Ressayre, Christine Dillmann
https://doi.org/10.1101/461947
Early and late flowering gene expression patterns in maize
Recommended by Tanja Pyhäjärvi based on reviews by Laura Shannon and 2 anonymous reviewers
Artificial selection experiments are key experiments in evolutionary biology. The demonstration that application of selective pressure across multiple generations results in heritable phenotypic changes is a tangible and reproducible proof of the evolution by natural selection.
Artificial selection experiments are used to evaluate the joint effects of selection on multiple traits, their genetic covariances and differences in responses in different environments. Most studies on artificial selection experiments report and base their analyses on phenotypic changes [1]. More recently, changes in allele frequency and other patterns of molecular genetic diversity have been used to identify genomic locations where selection has had an effect. However, so far the changes in gene expression have not been in the focus of artificial selection experiment studies (see [2] for an example though).
In plants, one of the most famous artificial selection experiments is the Illinois Corn Experiment where maize (Zea mays) is selected for oil and protein content [3], but in addition, similar experiments have been conducted also for other traits in maize. In Saclay divergent selection experiment [4] two maize inbred lines (F252 and MBS847) have been selected for early and late flowering for 13 generations, resulting in two week difference in flowering time.
In ”Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network ” [5] Maud Tenaillon and her coworkers study the gene expression differences among these two independently selected maize populations. Their experiments cover two years in field conditions and they use samples of shoot apical meristem at three different developmental stages: vegetative, transitioning and reproductive. They use RNA-seq transcriptome level differences and qRT-PCR for gene expression pattern investigation. The work is continuation to earlier genetic and phenotypic studies on the same material [4, 6].
The reviewers and I agree that dataset is unique and its major benefit is that it has been obtained from field conditions similar to those that species may face under natural setting during selection. Their tissue sampling is supported by flowering time phenotypic observations and covers the developmental transition stage, making a good effort to identify key transcriptional and phenotypic changes and their timing affected by selection.
Tenaillon et al. [5] identify more than 2000 genes that are differentially expressed among early and late flowering populations. Expectedly, they are enriched for known flowering time genes. As they point out, differential expression of thousands of genes does not mean that they all were independently affected by selection, but rather that the whole transcriptional network has shifted, possibly due to just few upstream or hub-genes. Also, the year-to-year variation had smaller effect in gene expression compared to developmental stage or genetic background, possibly indicating selection for stability across environmental fluctuation for such an important phenotype as flowering time.
Another noteworthy observation is that they find convergent patterns of transcriptional changes among the two selected lines. 115 genes expression patterns are shifted due to selection in both genetic backgrounds. This convergent pattern can be a result of either selection on standing variation or de novo mutations. The data does not allow testing which process is underlying the observed convergence. However, their results show that this is an interesting future question that can be addressed using genotype and gene expression data from the same ancestral and derived material and possibly their hybrids.
References
[1] Hill, W. G., & Caballero, A. (1992). Artificial selection experiments. Annual Review of Ecology and Systematics, 23(1), 287-310. doi: 10.1146/annurev.es.23.110192.001443
[2] Konczal, M., Babik, W., Radwan, J., Sadowska, E. T., & Koteja, P. (2015). Initial molecular-level response to artificial selection for increased aerobic metabolism occurs primarily through changes in gene expression. Molecular biology and evolution, 32(6), 1461-1473. doi: 10.1093/molbev/msv038
[3] Moose, S. P., Dudley, J. W., & Rocheford, T. R. (2004). Maize selection passes the century mark: a unique resource for 21st century genomics. Trends in plant science, 9(7), 358-364. doi: 10.1016/j.tplants.2004.05.005
[4] Durand, E., Tenaillon, M. I., Ridel, C., Coubriche, D., Jamin, P., Jouanne, S., Ressayre, A., Charcosset, A. and Dillmann, C. (2010). Standing variation and new mutations both contribute to a fast response to selection for flowering time in maize inbreds. BMC evolutionary biology, 10(1), 2. doi: 10.1186/1471-2148-10-2
[5] Tenaillon, M. I., Seddiki, K., Mollion, M., Le Guilloux, M., Marchadier, E., Ressayre, A. and Dillmann C. (2019). Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network. BioRxiv, 461947 ver. 5 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/461947
[6] Durand, E., Tenaillon, M. I., Raffoux, X., Thépot, S., Falque, M., Jamin, P., Bourgais A., Ressayre, A. and Dillmann, C. (2015). Dearth of polymorphism associated with a sustained response to selection for flowering time in maize. BMC evolutionary biology, 15(1), 103. doi: 10.1186/s12862-015-0382-5
| Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network | Maud Irène Tenaillon, Khawla Sedikki, Maeva Mollion, Martine Le Guilloux, Elodie Marchadier, Adrienne Ressayre, Christine Dillmann | <p>Artificial selection experiments are designed to investigate phenotypic evolution of complex traits and its genetic basis. Here we focused on flowering time, a trait of key importance for plant adaptation and life-cycle shifts. We undertook div... |  | Adaptation, Experimental Evolution, Expression Studies, Quantitative Genetics | Tanja Pyhäjärvi | | 2018-11-23 11:57:35 | View |







