
Latest recommendations

| Id | Title | Authors | Abstract | Picture▲ | Thematic fields | Recommender | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
02 May 2023
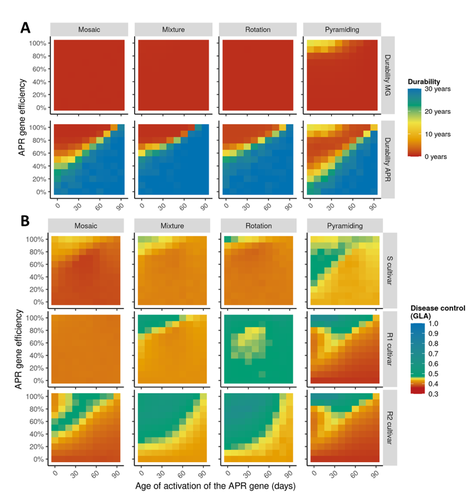
Durable resistance or efficient disease control? Adult Plant Resistance (APR) at the heart of the dilemmaPlant resistance to pathogens: just you wait?Recommended by Timothée Poisot based on reviews by Jean-Paul Soularue and 1 anonymous reviewerIn this preprint, Rimbaud et al. (2023) examine whether Adult Plant Resistance (APR), where plants delay their response to pathogens, is a viable alternative when the solution to evolve complete resistance from the seedling stage exists. At first glance, delaying resistance seems like a counter-intuitive strategy, unless it can result in a weaker selection of the pathogen, and therefore slow down its adaption to plant resistance. The approach of Rimbaud et al. is to incorporate as much of the mechanisms as possible into a model. By accounting for explicit spatio-temporal dynamics, stochasticity, and the coupling between demography and population genetics, to simulate an agricultural landscape, they reach a nuanced conclusion. Weaker and delayed activation of genes that confer APR does indeed reduce the selection pressure acting on the pathogen, at the cost of overall less effective protection. The alternative strategy of rapid or complete activation of these genes, although it results in better results in defending against the pathogen, is at risk of being overcome because it introduces a stronger selection pressure. One important feature of this work is that it accounts for agricultural practices. The landscape that is simulated can account for monoculture, mosaic cultures, mixed cultures, and rotations of crops (with different strategies for resistance). This introduces an interesting element to the conclusion: that human practices will have an impact on the selection pressures acting within the system. Perhaps the most striking result is that, for the plants, it might be more beneficial to bear the cost of a wild-type pathogen that can benefit from delayed activation of resistance, and therefore exclude the more virulent strains by simply being there first, and essentially buying the plant some time before it activates its resistance more completely. When the landscape is aggregated, even wild-type pathogens can cause severe epidemics; increasing fragmentation, because it enables connectivity between patches of plants with different strategies, allows pathogens to move across cultivars, and reduces the epidemic risk on susceptible plants. These results should encourage scaling up the perspective on APR, and indeed Rimbaud et al. adopt a landscape-scale perspective, to show that APR genes and genes conferring more complete resistance early on can have synergistic effects. This is, again, both an interesting result for evolutionary biologists, but also a useful way to prioritize different crop management strategies over large spatial scales. References Rimbaud, Loup, et al. Durable Resistance or Efficient Disease Control? Adult Plant Resistance (APR) at the Heart of the Dilemma. 2023. bioRxiv, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.08.30.505787 | Durable resistance or efficient disease control? Adult Plant Resistance (APR) at the heart of the dilemma | Loup Rimbaud, Julien Papaïx, Jean-François Rey, Benoît Moury, Luke G. Barrett, Peter H. Thrall | <p style="text-align: justify;">Adult plant resistance (APR) is an incomplete and delayed protection of plants against pathogens. At first glance, such resistance should be less efficient than classical major-effect resistance genes, which confer ... | | Adaptation, Evolutionary Applications, Evolutionary Epidemiology | Timothée Poisot | 2022-09-02 16:36:32 | ||
13 Dec 2018
Separate the wheat from the chaff: genomic analysis of local adaptation in the red coral Corallium rubrumPros and Cons of local adaptation scansRecommended by Guillaume Achaz based on reviews by Lucas Gonçalves da Silva and 1 anonymous reviewerThe preprint by Pratlong et al. [1] is a well thought quest for genomic regions involved in local adaptation to depth in a species a red coral living the Mediterranean Sea. It first describes a pattern of structuration and then attempts to find candidate genes involved in local adaptation by contrasting deep with shallow populations. Although the pattern of structuration is clear and meaningful, the candidate genomic regions involved in local adaptation remain to be confirmed. Two external reviewers and myself found this preprint particularly interesting regarding the right-mindedness of the authors in front of the difficulties they encounter during their experiments. The discussions on the pros and cons of the approach are very sound and can be easily exported to a large number of studies that hunt for local adaptation. In this sense, the lessons one can learn by reading this well documented manuscript are certainly valuable for a wide range of evolutionary biologists. References [1] Pratlong, M., Haguenauer, A., Brener, K., Mitta, G., Toulza, E., Garrabou, J., Bensoussan, N., Pontarotti P., & Aurelle, D. (2018). Separate the wheat from the chaff: genomic scan for local adaptation in the red coral Corallium rubrum. bioRxiv, 306456, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/306456 | Separate the wheat from the chaff: genomic analysis of local adaptation in the red coral Corallium rubrum | Pratlong M, Haguenauer A, Brener K, Mitta G, Toulza E, Garrabou J, Bensoussan N, Pontarotti P, Aurelle D | <p>Genomic data allow an in-depth and renewed study of local adaptation. The red coral (Corallium rubrum, Cnidaria) is a highly genetically structured species and a promising model for the study of adaptive processes along an environmental gradien... | | Adaptation, Population Genetics / Genomics | Guillaume Achaz | 2018-04-24 11:27:40 | ||
25 Mar 2019

The joint evolution of lifespan and self-fertilisationEvolution of selfing & lifespan 2.0Recommended by Thomas Bataillon based on reviews by 2 anonymous reviewersFlowering plants display a staggering diversity of both mating systems and life histories, ranging from almost exclusively selfers to obligate outcrossers, very short-lived annual herbs to super long lived trees. One pervasive pattern that has attracted considerable attention is the tight correlation that is found between mating systems and lifespan [1]. Until recently, theoretical explanations for these patterns have relied on static models exploring the consequences of several non-mutually exclusive important process: levels of inbreeding depression and ability to successfully were center stage. This make sense: successful colonization after long‐distance dispersal is far more likely to happen for self‐compatible than for self‐incompatible individuals in a sexually reproducing species. Furthermore, inbreeding depression (essentially a genetically driven phenomenon) and reproductive insurance are expected to shape the evolution of both mating system and lifespan. References | The joint evolution of lifespan and self-fertilisation | Thomas Lesaffre, Sylvain Billiard | <p>In Angiosperms, there exists a strong association between mating system and lifespan. Most self-fertilising species are short-lived and most predominant or obligate outcrossers are long-lived. This association is generally explained by the infl... | | Evolutionary Theory, Life History, Reproduction and Sex | Thomas Bataillon | 2018-09-19 10:03:51 | ||
16 Dec 2016
POSTPRINT
Spatiotemporal microbial evolution on antibiotic landscapesA poster child for experimental evolutionRecommended by Daniel Rozen and Arjan de VisserEvolution is usually studied via two distinct approaches: by inferring evolutionary processes from relatedness patterns among living species or by observing evolution in action in the laboratory or field. A recent study by Baym and colleagues in Science [1] has now combined these approaches by taking advantage of the pattern left behind by spatially evolving bacterial populations. Evolution is often considered too slow to see, and can only be inferred by studying patterns of relatedness using phylogenetic trees. Increasingly, however, researchers are moving nature into the lab and watching as evolution unfolds under their noses. The field of experimental evolution follows evolutionary change in the laboratory over 10s to 1000s of generations, yielding insights into bacterial, viral, plant, or fly evolution (among many other species) that are simply not possible in the field. Yet, as powerful as experimental evolution is, it lacks a posterchild. There is no Galapagos finch radiation, nor a stunning series of cichlids to showcase to our students to pique their interests. Let’s face it, E. coli is no stickleback! And while practitioners of experimental evolution can explain the virtues of examining 60,000 generations of bacterial evolution in action, appreciating this nevertheless requires a level of insight and imagination that often eludes students, who need to see “it” to get it. Enter MEGA, an idea and a film that could become the new face of experimental evolution. It replaces big numbers of generations or images of scientists, with an actual picture of the scientific result. MEGA, or Microbial Evolution and Growth Arena, is essentially an enormous petri dish and is the brainchild of Michael Baym, Tami Leiberman and their colleagues in Roy Kishony’s lab at Technion Israel Institute of Technology and Harvard Medical School. The idea of MEGA is to allow bacteria to swim over a spatially defined landscape while adapting to the local conditions, in this case antibiotics. When bacteria are inoculated onto one end of the plate they consume resources while swarming forward from the plate edge. In a few days, the bacteria grow into an area with antibiotics to which they are susceptible. This stops growth until a mutation arises that permits the bacteria to jump this hurdle, after which growth proceeds until the next hurdle of a 10-fold higher antibiotic concentration, and so on. By this simple approach, Baym et al. [1] evolved E. coli that were nearly 105-fold more resistant to two different antibiotics in just over 10 days. In addition, they identified the mutations that were required for these changes, showed that mutations conferring smaller benefits were required before bacteria could evolve maximal resistance, observed changes to the mutation rate, and demonstrated the importance of spatial structure in constraining adaptation. For one thing, the rate of resistance evolution is impressive, and also quite scary given the mounting threat of antibiotic-resistant pathogens. However, MEGA also offers a uniquely visual insight into evolutionary change. By taking successive images of the MEGA plate, the group was able to watch the bacteria move, get trapped because of their susceptibility to the antibiotic, and then get past these traps as new mutations emerged that increased resistance. Each transition showcases evolution in real time. In addition, by leaving a spatial pattern of evolutionary steps behind, the MEGA plate offers unique opportunities to thoroughly investigate these steps when the experiment is finished. For instance, subsequent steps in mutational pathways can be characterized, but also their effects on fitness can be quantified in situ by measuring changes in survival and reproduction. This new method is undoubtedly a boon to the field of experimental evolution and offers endless opportunities for experimental elaboration. Perhaps of equal importance, MEGA is a tool that is portable to the classroom and to the public at large. Don’t believe in evolution? Watch this. You only have time for a short internship or lab practical? No problem. Don’t worry much about antibiotic resistance? Check this out. Like the best experimental tools, MEGA is simple but allows for complicated insights. And even if it is less charismatic than a finch, it still allows for the kinds of “gee-whiz” insights that will get students hooked on evolutionary biology. Reference [1] Baym M, Lieberman TD, Kelsic ED, Chait R, Gross R, Yelin I, Kishony R. 2016. Spatiotemporal microbial evolution on antibiotic landscapes. Science 353:1147-1151. doi: 10.1126/science.aag0822 | Spatiotemporal microbial evolution on antibiotic landscapes | Baym M, Lieberman TD, Kelsic ED, Chait R, Gross R, Yelin I, Kishony R | A key aspect of bacterial survival is the ability to evolve while migrating across spatially varying environmental challenges. Laboratory experiments, however, often study evolution in well-mixed systems. Here, we introduce an experimental device,... | | Adaptation, Evolutionary Applications, Experimental Evolution | Daniel Rozen | 2016-12-14 14:26:06 | ||
19 Mar 2018

Natural selection on plasticity of thermal traits in a highly seasonal environmentIs thermal plasticity itself shaped by natural selection? An assessment with desert frogsRecommended by Wolf Blanckenhorn based on reviews by Dries Bonte, Wolf Blanckenhorn and Nadia Aubin-HorthIt is well known that climatic factors – most notably temperature, season length, insolation and humidity – shape the thermal niche of organisms on earth through the action of natural selection. But how is this achieved precisely? Much of thermal tolerance is actually mediated by phenotypic plasticity (as opposed to genetic adaptation). A prominent expectation is that environments with greater (daily and/or annual) thermal variability select for greater plasticity, i.e. better acclimation capacity. Thus, plasticity might be selected per se. A Chilean group around Leonardo Bacigalupe assessed natural selection in the wild in one marginal (and extreme) population of the four-eyed frog Pleurodema thaul (Anura: Leptodactylidae) in an isolated oasis in the Atacama Desert, permitting estimation of mortality without much potential of confounding it with migration [1]. Several thermal traits were considered: CTmax – the critical maximal temperature; CTmin – the critical minimum temperature; Tpref – preferred temperature; Q10 – thermal sensitivity of metabolism; and body mass. Animals were captured in the wild and subsequently assessed for thermal traits in the laboratory at two acclimation temperatures (10° & 20°C), defining the plasticity in all traits as the difference between the traits at the two acclimation temperatures. Thereafter the animals were released again in their natural habitat and their survival was monitored over the subsequent 1.5 years, covering two breeding seasons, to estimate viability selection in the wild. The authors found and conclude that, aside from larger body size increasing survival (an unsurprising result), plasticity does not seem to be systematically selected directly, while some of the individual traits show weak signs of selection. Despite limited sample size (ca. 80 frogs) investigated in only one marginal but very seasonal population, this study is interesting because selection on plasticity in physiological thermal traits, as opposed to selection on the thermal traits themselves, is rarely investigated. The study thus also addressed the old but important question of whether plasticity (i.e. CTmax-CTmin) is a trait by itself or an epiphenomenon defined by the actual traits (CTmax and CTmin) [2-5]. Given negative results, the main question could not be ultimately solved here, so more similar studies should be performed. References [1] Bacigalupe LD, Gaitan-Espitia, JD, Barria AM, Gonzalez-Mendez A, Ruiz-Aravena M, Trinder M & Sinervo B. 2018. Natural selection on plasticity of thermal traits in a highly seasonal environment. bioRxiv 191825, ver. 5 peer-reviewed by Peer Community In Evolutionary Biology. doi: 10.1101/191825 | Natural selection on plasticity of thermal traits in a highly seasonal environment | Leonardo Bacigalupe, Juan Diego Gaitan-Espitia, Aura M Barria, Avia Gonzalez-Mendez, Manuel Ruiz-Aravena, Mark Trinder, Barry Sinervo | <p>For ectothermic species with broad geographical distributions, latitudinal/altitudinal variation in environmental temperatures (averages and extremes) are expected to shape the evolution of physiological tolerances and the acclimation capacity ... | | Adaptation, Evolutionary Ecology, Phenotypic Plasticity | Wolf Blanckenhorn | 2017-09-22 23:17:40 | ||
11 Apr 2023
Facultative parthenogenesis: a transient state in transitions between sex and obligate asexuality in stick insects?Facultative parthenogenesis and transitions from sexual to asexual reproductionRecommended by Trine Bilde based on reviews by 3 anonymous reviewers based on reviews by 3 anonymous reviewers
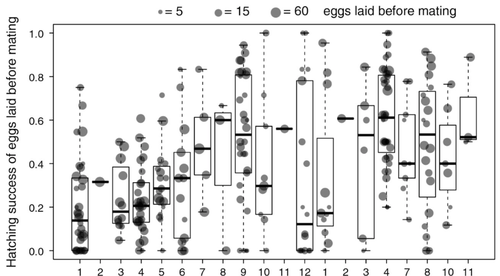
Despite a vast array of ways in which organisms can reproduce (Bell, 1982), most animals engage in sexual reproduction (Otto & Lenormand, 2002). A fascinating alternative to sex is parthenogenesis, where offspring are produced asexually from a gamete, typically the egg, without receiving genetic material from another gamete (Simon, Delmotte, Rispe, & Crease, 2003). One of the long-standing questions in the field is why parthenogenesis is not more widespread, given the costs associated with sex (Otto & Lenormand, 2002). Natural populations of most species appear to be reproducing either sexually or parthenogenetically, even if a species can employ both reproductive modes (Larose et al 2023). Larose et al (2023) highlight the conundrum in this pattern, as organisms that are capable of employing parthenogenesis facultatively would be able to gain the benefits of both modes of reproduction. Why then, is facultative parthenogenesis not more common? Larose et al (2023) propose that constraints on being efficient in both sexual and asexual reproduction could cause a trade-off between reproductive modes that favours an obligate strategy of either sex or no sex. This would provide an explanation for why facultative parthenogenesis is rare. Timema stick insects provide an excellent system to investigate reproductive strategies, as some species have parthenogenetic females, while other species are sexual, and they show repeated transitions from sexual reproduction to obligate parthenogenesis (Schwander & Crespi, 2009). The authors performed comprehensive and complementary studies in a recently discovered species T. douglasi, in which populations show both modes of reproduction, with some populations consisting only of females and others showing equal proportions of males and females. The sex ratio varied significantly, with the proportion of females ranging between 43-100% across 29 populations. These populations form a monophyletic clade with clustering into three genetic lineages and only a few cases of admixture. Females from all populations were capable of producing unfertilized eggs, but the hatching success varied hugely among populations and lineages (3-100%). Parthenogenetically produced offspring were homozygous, showing that parthenogenesis causes a complete loss of heterozygosity in a single generation. After producing eggs as virgins, females were mated to assess the capacity to also reproduce sexually, and fertilization increased the hatching success of eggs in two lineages. In one lineage, in which the hatching success of unfertilized eggs is similar to that of other sexually reproducing Timema species, fertilization reduced egg-hatching success, indicating a trade-off between reproductive modes with parthenogenetic reproduction performing best. Approximately 58% of the offspring produced after mating were fertilized, demonstrating the capacity of females to reproduce parthenogenetically also after mating has occurred, however with huge variation among individuals. This wonderful and meticulously performed study produces strong and complementary evidence for facultative parthenogenesis in T. douglasi populations. The study shows large variation in how reproductive mode is employed, supporting the existence of a trade-off between sexual and parthenogenetic reproduction. This might be an example of an ongoing transition from sexual to asexual reproduction, which indicates that obligate parthenogenesis may derive via transient facultative parthenogenesis. REFERENCES Bell, G. (1982) The Masterpiece of Nature: The Evolution and Genetics of Sexuality. University of California Press. 635 p. Otto, S. P., & Lenormand, T. (2002). Resolving the paradox of sex and recombination. Nature Reviews Genetics, 3(4), 252-261. https://doi.org/10.1038/nrg761 Schwander, T., & Crespi, B. J. (2009). Multiple direct transitions from sexual reproduction to apomictic parthenogenesis in Timema stick insects. Evolution, 63(1), 84-103. Simon, J.-C., Delmotte, F., Rispe, C., & Crease, T. (2003). Phylogenetic relationships between parthenogens and their sexual relatives: the possible routes to parthenogenesis in animals. Biological Journal of the Linnean Society, 79(1), 151-163. https://doi.org/10.1046/j.1095-8312.2003.00175.x Larose, C., Lavanchy, G., Freitas, S., Parker, D.J., Schwander, T. (2023) Facultative parthenogenesis: a transient state in transitions between sex and obligate asexuality in stick insects? bioRxiv, 2022.03.25.485836, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.03.25.485836 | Facultative parthenogenesis: a transient state in transitions between sex and obligate asexuality in stick insects? | Chloé Larose, Guillaume Lavanchy, Susana Freitas, Darren J. Parker, Tanja Schwander | <p>Transitions from obligate sex to obligate parthenogenesis have occurred repeatedly across the tree of life. Whether these transitions occur abruptly or via a transient phase of facultative parthenogenesis is rarely known. We discovered and char... | | Reproduction and Sex | Trine Bilde | 2022-05-20 10:41:13 | ||
21 Feb 2023
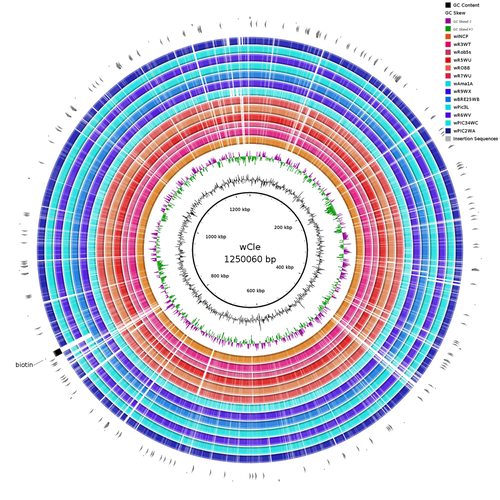
Wolbachia genomics reveals a potential for a nutrition-based symbiosis in blood-sucking Triatomine bugsNutritional symbioses in triatomines: who is playing?Recommended by Natacha Kremer based on reviews by Alejandro Manzano Marín and Olivier DuronNearly 8 million people are suffering from Chagas disease in the Americas. The etiological agent, Trypanosoma cruzi, is mainly transmitted by triatomine bugs, also known as kissing or vampire bugs, which suck blood and transmit the parasite through their feces. Among these triatomine species, Rhodnius prolixus is considered the main vector, and many studies have focused on characterizing its biology, physiology, ecology and evolution. Interestingly, given that Rhodnius species feed almost exclusively on blood, their diet is unbalanced, and the insects can lack nutrients and vitamins that they cannot synthetize themself, such as B-vitamins. In all insects feeding exclusively on blood, symbioses with microbes producing B-vitamins (mainly biotin, riboflavin and folate) have been widely described (see review in Duron and Gottlieb 2020) and are critical for insect development and reproduction. These co-evolved relationships between blood feeders and nutritional symbionts could now be considered to develop new control methods, by targeting the ‘Achille’s heel’ of the symbiotic association (i.e., transfer of nutrient and / or control of nutritional symbiont density). But for this, it is necessary to better characterize the relationships between triatomines and their symbionts. R. prolixus is known to be associated with several symbionts. The extracellular gut symbiont Rhodococcus rhodnii, which reaches high bacterial densities and is almost fixed in R. prolixus populations, appears to be a nutritional symbiont under many blood sources. This symbiont can provide B-vitamins such as biotin (B7), niacin (B3), thiamin (B1), pyridoxin (B6) or riboflavin (B2) and can play an important role in the development and the reproduction of R. prolixus (Pachebat et al. (2013) and see review in Salcedo-Porras et al. (2020)). This symbiont is orally acquired through egg smearing, ensuring the fidelity of transmission of the symbiont from mother to offspring. However, as recently highlighted by Tobias et al. (2020) and Gilliland et al. (2022), other gut microbes could also participate to the provision of B-vitamins, and R. rhodnii could additionally provide metabolites (other than B-vitamins) increasing bug fitness. In the study from Filée et al., the authors focused on Wolbachia, an intracellular, maternally inherited bacterium, known to be a nutritional symbiont in other blood-sucking insects such as bedbugs (Nikoh et al. 2014), and its potential role in vitamin provision in triatomine bugs. After screening 17 different triatomine species from the 3 phylogenetic groups prolixus, pallescens and pictipes, they first show that Wolbachia symbionts are widely distributed in the different Rhodnius species. Contrary to R. rhodnii that were detected in all samples, Wolbachia prevalence was patchy and rarely fixed. The authors then sequenced, assembled, and compared 13 Wolbachia genomes from the infected Rhodnius species. They showed that all Wolbachia are phylogenetically positioned in the supergroup F that contains wCle (the Wolbachia from bedbugs). In addition, 8 Wolbachia strains (out of 12) encode a biotin operon under strong purifying selection, suggesting the preservation of the biological function and the metabolic potential of Wolbachia to supplement biotin in their Rhodnius host. From the study of insect genomes, the authors also evidenced several horizontal transfers of genes from Wolbachia to Rhodnius genomes, which suggests a complex evolutionary interplay between vampire bugs and their intracellular symbiont. This nice piece of work thus provides valuable information to the fields of multiple partners / nutritional symbioses and Wolbachia research. Dual symbioses described in insects feeding on unbalanced diets generally highlight a certain complementarity between symbionts that ensure the whole nutritional complementation. The study presented by Filée et al. leads rather to consider the impact of multiple symbionts with different lifestyles and transmission modes in the provision of a specific nutritional benefit (here, biotin). Because of the low prevalence of Wolbachia in certain species, a “ménage à trois” scenario would rather be replaced by an “open couple”, where the host relationship with new symbiotic partners (more or less stable at the evolutionary timescale) could provide benefits in certain ecological situations. The results also support the potential for Wolbachia to evolve rapidly along a continuum between parasitism and mutualism, by acquiring operons encoding critical pathways of vitamin biosynthesis. References Duron O. and Gottlieb Y. (2020) Convergence of Nutritional Symbioses in Obligate Blood Feeders. Trends in Parasitology 36(10):816-825. https://doi.org/10.1016/j.pt.2020.07.007 Filée J., Agésilas-Lequeux K., Lacquehay L., Bérenger J.-M., Dupont L., Mendonça V., Aristeu da Rosa J. and Harry M. (2023) Wolbachia genomics reveals a potential for a nutrition-based symbiosis in blood-sucking Triatomine bugs. bioRxiv, 2022.09.06.506778, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.09.06.506778 Gilliland C.A. et al. (2022) Using axenic and gnotobiotic insects to examine the role of different microbes on the development and reproduction of the kissing bug Rhodnius prolixus (Hemiptera: Reduviidae). Molecular Ecology. https://doi.org/10.1111/mec.16800 Nikoh et al. (2014) Evolutionary origin of insect–Wolbachia nutritional mutualism. PNAS. 111(28):10257-10262. https://doi.org/10.1073/pnas.1409284111 Pachebat, J.A. et al. (2013). Draft genome sequence of Rhodococcus rhodnii strain LMG5362, a symbiont of Rhodnius prolixus (Hemiptera, Reduviidae, Triatominae), the principle vector of Trypanosoma cruzi. Genome Announc. 1(3):e00329-13. https://doi.org/10.1128/genomea.00329-13 Salcedo-Porras N., et al. (2020). The role of bacterial symbionts in Triatomines: an evolutionary perspective. Microorganisms. 8:1438. https://doi.org/10.3390%2Fmicroorganisms8091438 Tobias N.J., Eberhard F.E., Guarneri A.A. (2020) Enzymatic biosynthesis of B-complex vitamins is supplied by diverse microbiota in the Rhodnius prolixus anterior midgut following Trypanosoma cruzi infection. Computational and Structural Biotechnology Journal. 3395-3401. https://doi.org/10.1016/j.csbj.2020.10.031 | Wolbachia genomics reveals a potential for a nutrition-based symbiosis in blood-sucking Triatomine bugs | Jonathan Filée, Kenny Agésilas-Lequeux, Laurie Lacquehay, Jean Michel Bérenger, Lise Dupont, Vagner Mendonça, João Aristeu da Rosa, Myriam Harry | <p>The nutritional symbiosis promoted by bacteria is a key determinant for adaptation and evolution of many insect lineages. A complex form of nutritional mutualism that arose in blood-sucking insects critically depends on diverse bacterial symbio... | | Genome Evolution, Phylogenetics / Phylogenomics, Species interactions | Natacha Kremer | Alejandro Manzano Marín | 2022-09-13 17:36:46 | |
28 Mar 2019
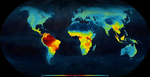
Ancient tropical extinctions contributed to the latitudinal diversity gradientOne (more) step towards a dynamic view of the Latitudinal Diversity GradientRecommended by Joaquín Hortal and Juan Arroyo based on reviews by Juan Arroyo, Joaquín Hortal, Arne Mooers, Joaquin Calatayud and 2 anonymous reviewers
The Latitudinal Diversity Gradient (LDG) has fascinated natural historians, ecologists and evolutionary biologists ever since [1] described it about 200 years ago [2]. Despite such interest, agreement on the origin and nature of this gradient has been elusive. Several tens of hypotheses and models have been put forward as explanations for the LDG [2-3], that can be grouped in ecological, evolutionary and historical explanations [4] (see also [5]). These explanations can be reduced to no less than 26 hypotheses, which account for variations in ecological limits for the establishment of progressively larger assemblages, diversification rates, and time for species accumulation [5]. Besides that, although in general the tropics hold more species, different taxa show different shapes and rates of spatial variation [6], and a considerable number of groups show reverse patterns, with richer assemblages in cold temperate regions (see e.g. [7-9]). References | Ancient tropical extinctions contributed to the latitudinal diversity gradient | Andrea S. Meseguer, Fabien Condamine | <p>Biodiversity currently peaks at the equator, decreasing toward the poles. Growing fossil evidence suggest that this hump-shaped latitudinal diversity gradient (LDG) has not been persistent through time, with similar species diversity across lat... | | Evolutionary Dynamics, Evolutionary Ecology, Macroevolution, Paleontology, Phylogenetics / Phylogenomics, Phylogeography & Biogeography | Joaquín Hortal | 2017-12-20 14:58:01 | ||
23 Nov 2020

Wolbachia and host intrinsic reproductive barriers contribute additively to post-mating isolation in spider mitesSpeciation in spider mites: disentangling the roles of Wolbachia-induced vs. nuclear mating incompatibilitiesRecommended by Jan Engelstaedter based on reviews by Wolfgang Miller and 1 anonymous reviewerCytoplasmic incompatibility (CI) is a mating incompatibility that is induced by maternally inherited endosymbionts in many arthropods. These endosymbionts include, most famously, the alpha-proteobacterium Wolbachia pipientis (Yen & Barr 1971; Werren et al. 2008) but also the Bacteroidetes bacterium Cardinium hertigii (Zchori-Fein et al. 2001), a gamma-proteobacterium of the genus Rickettsiella (Rosenwald et al. 2020) and another, as yet undescribed alpha-proteobacterium (Takano et al. 2017). CI manifests as embryonic mortality in crosses between infected males and females that are uninfected or infected with a different strain, whereas embryos develop normally in all other crosses. This phenotype may enable the endosymbionts to spread rapidly within their host population. Exploiting this, CI-inducing Wolbachia are being harnessed to control insect-borne diseases (e.g., O'Neill 2018). Much progress elucidating the genetic basis and developmental mechanism of CI has been made in recent years, but many open questions remain (Shropshire et al. 2020). References Bordenstein, S. R., O'Hara, F. P., and Werren, J. H. (2001). Wolbachia-induced incompatibility precedes other hybrid incompatibilities in Nasonia. Nature, 409(6821), 707-710. doi: https://doi.org/10.1038/35055543 | Wolbachia and host intrinsic reproductive barriers contribute additively to post-mating isolation in spider mites | Miguel A. Cruz, Sara Magalhães, Élio Sucena, Flore Zélé | <p>Wolbachia are widespread maternally-inherited bacteria suggested to play a role in arthropod host speciation through induction of cytoplasmic incompatibility, but this hypothesis remains controversial. Most studies addressing Wolbachia-induced ... | | Evolutionary Ecology, Hybridization / Introgression, Life History, Reproduction and Sex, Speciation, Species interactions | Jan Engelstaedter | 2020-07-09 10:18:28 | ||
09 Dec 2019
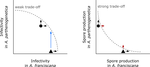
Trait-specific trade-offs prevent niche expansion in two parasitesTrade-offs in fitness components and ecological source-sink dynamics affect host specialisation in two parasites of Artemia shrimpsRecommended by Frédéric Guillaume based on reviews by Anne Duplouy, Seth Barribeau and Cindy Gidoin
Ecological specialisation, especially among parasites infecting a set of host species, is ubiquitous in nature. Host specialisation can be understood as resulting from trade-offs in parasite infectivity, virulence and growth. However, it is not well understood how variation in these trade-offs shapes the overall fitness trade-off a parasite faces when adapting to multiple hosts. For instance, it is not clear whether a strong trade-off in one fitness component may sufficiently constrain the evolution of a generalist parasite despite weak trade-offs in other components. A second mechanism explaining variation in specialisation among species is habitat availability and quality. Rare habitats or habitats that act as ecological sinks will not allow a species to persist and adapt, preventing a generalist phenotype to evolve. Understanding the prevalence of those mechanisms in natural systems is crucial to understand the emergence and maintenance of host specialisation, and biodiversity in general. References [1] Lievens, E.J.P., Michalakis, Y. and Lenormand, T. (2019). Trait-specific trade-offs prevent niche expansion in two parasites. bioRxiv, 621581, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/621581 | Trait-specific trade-offs prevent niche expansion in two parasites | Eva JP Lievens, Yannis Michalakis, Thomas Lenormand | <p>The evolution of host specialization has been studied intensively, yet it is still often difficult to determine why parasites do not evolve broader niches – in particular when the available hosts are closely related and ecologically similar. He... | | Adaptation, Evolutionary Ecology, Evolutionary Epidemiology, Experimental Evolution, Life History, Species interactions | Frédéric Guillaume | 2019-05-13 13:44:34 |




