Transcriptional differences between the two host strains of Spodoptera frugiperda (Lepidoptera: Noctuidae)
Marion Orsucci, Yves Moné, Philippe Audiot, Sylvie Gimenez, Sandra Nhim, Rima Naït-Saïdi, Marie Frayssinet, Guillaume Dumont, Jean-Paul Boudon, Marin Vabre, Stéphanie Rialle, Rachid Koual, Gael J. Kergoat, Rodney N. Nagoshi, Robert L. Meagher, Emmanuelle d'Alencon, Nicolas Nègre
https://doi.org/10.1101/263186
Speciation through selection on mitochondrial genes?
Recommended by Astrid Groot based on reviews by Heiko Vogel and Sabine Haenniger
Whether speciation through ecological specialization occurs has been a thriving research area ever since Mayr (1942) stated this to play a central role. In herbivorous insects, ecological specialization is most likely to happen through host plant differentiation (Funk et al. 2002). Therefore, after Dorothy Pashley had identified two host strains in the Fall armyworm (FAW), Spodoptera frugiperda, in 1988 (Pashley 1988), researchers have been trying to decipher the evolutionary history of these strains, as this seems to be a model species in which speciation is currently occurring through host plant specialization. Even though FAW is a generalist, feeding on many different host plant species (Pogue 2002) and a devastating pest in many crops, Pashley identified a so-called corn strain and a so-called rice strain in Puerto Rico. Genetically, these strains were found to differ mostly in an esterase, although later studies showed additional genetic differences and markers, mostly in the mitochondrial COI and the nuclear TPI. Recent genomic studies showed that the two strains are overall so genetically different (2% of their genome being different) that these two strains could better be called different species (Kergoat et al. 2012). So far, the most consistent differences between the strains have been their timing of mating activities at night (Schoefl et al. 2009, 2011; Haenniger et al. 2019) and hybrid incompatibilities (Dumas et al. 2015; Kost et al. 2016). Whether and to what extent host plant preference or performance contributed to the differentiation of these sympatrically occurring strains has remained unclear.
In the current study, Orsucci et al. (2020) performed oviposition assays and reciprocal transplant experiments with both strains to measure fitness effects, in combination with a comprehensive RNAseq experiment, in which not only lab reared larvae were analysed, but also field-collected larvae. When testing preference and performance on the two host plants corn and rice, the authors did not find consistent fitness differences between the two strains, with both strains performing less on rice plants, although larvae from the corn strain survived more on corn plants than those from the rice strain. These results mostly confirm findings of a number of investigations over the past 30 years, where no consistent differences on the two host plants were observed (reviewed in Groot et al. 2016). However, the RNAseq experiments did show some striking differences between the two strains, especially in the reciprocally transplanted larvae, where both strains had been reared on rice or on corn plants for one generation: both strains showed transcriptional responses that correspond to their respective putative host plants, i.e. overexpression of genes involved in digestion and metabolic activity, and underexpression of genes involved in detoxification, in the corn strain on corn and in the rice strain on rice. Interestingly, similar sets of genes were found to be overexpressed in the field-collected larvae with which a RNAseq experiment was conducted as well.
The most interesting result of the study performed by Orsucci et al. (2020) is the underexpression in the corn strain of so-called numts, small genomic sequences that corresponded to fragments of the mitochondrial COI and COIII. These two numts were differentially expressed in the two strains in all RNAseq experiments analysed. This result coincides with the fact that the COI is one of the main diagnostic markers to distinguish these two strains. The authors suggestion that a difference in energy production between these two strains may be linked to a shift in host plant preference matches their finding that rice plants seem to be less suitable host plants than corn plants. However, as the lower suitability of rice plants was true for both strains, it remains unclear whether and how this difference could be linked to possible host plant differentiation between the strains. The authors also suggest that COI and potentially other mitochondrial genes may be the original target of selection between these two strains. This is especially interesting in light of the fact that field-collected larvae have frequently been found to have a rice strain mitochondrial genotype and a corn strain nuclear genotype, also in this study, while in the lab (female rice strain x male corn strain) hybrid females (i.e. females with a rice strain mitochondrial genotype and a corn strain nuclear genotype) are behaviorally sterile (Kost et al. 2016). Whether and how selection on mitochondrial genes or on mitonuclear interactions has indeed affected the evolution of these strains in the New world, and will affect the evolution of FAW in newly invaded habitats in the Old world, including Asia and Australia – where, so far, only corn strain and (female rice strain x male corn strain) hybrids have been found (Nagoshi 2019), will be a challenging research question for the coming years.
References
[1] Dumas, P. et al. (2015). Spodoptera frugiperda (Lepidoptera: Noctuidae) host-plant variants: two host strains or two distinct species?. Genetica, 143(3), 305-316. doi: 10.1007/s10709-015-9829-2
[2] Funk, D. J., Filchak, K. E. and Feder J. L. (2002) Herbivorous insects: model systems for the comparative study of speciation ecology. In: Etges W.J., Noor M.A.F. (eds) Genetics of Mate Choice: From Sexual Selection to Sexual Isolation. Contemporary Issues in Genetics and Evolution, vol 9. Springer, Dordrecht. doi: 10.1007/978-94-010-0265-3_10
[3] Groot, A. T., Unbehend, M., Hänniger, S., Juárez, M. L., Kost, S. and Heckel D. G.(2016) Evolution of reproductive isolation of Spodoptera frugiperda. In Allison, J. and Cardé, R. (eds) Sexual communication in moths. Chapter 20: 291-300.
[4] Hänniger, S. et al. (2017). Genetic basis of allochronic differentiation in the fall armyworm. BMC evolutionary biology, 17(1), 68. doi: 10.1186/s12862-017-0911-5
[5] Kost, S., Heckel, D. G., Yoshido, A., Marec, F., and Groot, A. T. (2016). AZ‐linked sterility locus causes sexual abstinence in hybrid females and facilitates speciation in Spodoptera frugiperda. Evolution, 70(6), 1418-1427. doi: 10.1111/evo.12940
[6] Mayr, E. (1942) Systematics and the origin of species. Columbia University Press, New York.
[7] Nagoshi, R. N. (2019). Evidence that a major subpopulation of fall armyworm found in the Western Hemisphere is rare or absent in Africa, which may limit the range of crops at risk of infestation. PloS one, 14(4). doi: 10.1371/journal.pone.0208966
[8] Orsucci, M., Moné, Y., Audiot, P., Gimenez, S., Nhim, S., Naït-Saïdi, R., Frayssinet, M., Dumont, G., Boudon, J.-P., Vabre, M., Rialle, S., Koual, R., Kergoat, G. J., Nagoshi, R. N., Meagher, R. L., d’Alençon, E. and Nègre N. (2020) Transcriptional differences between the two host strains of Spodoptera frugiperda (Lepidoptera: Noctuidae). bioRxiv, 263186, ver. 2 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/263186
[9] Pashley, D. P. (1988) Current Status of Fall Armyworm Host Strains. Florida Entomologist 71 (3): 227–34. doi: 10.2307/3495425
[10] Pogue, M. (2002). A World Revision of the Genus Spodoptera Guenée (Lepidoptera: Noctuidae). American Entomological Society.
[11] Schöfl, G., Heckel, D. G., and Groot, A. T. (2009). Time‐shifted reproductive behaviours among fall armyworm (Noctuidae: Spodoptera frugiperda) host strains: evidence for differing modes of inheritance. Journal of Evolutionary Biology, 22(7), 1447-1459. doi: 10.1111/j.1420-9101.2009.01759.x
[12] Schöfl, G., Dill, A., Heckel, D. G., and Groot, A. T. (2011). Allochronic separation versus mate choice: nonrandom patterns of mating between fall armyworm host strains. The American Naturalist, 177(4), 470-485. doi: 10.1086/658904
| Transcriptional differences between the two host strains of Spodoptera frugiperda (Lepidoptera: Noctuidae) | Marion Orsucci, Yves Moné, Philippe Audiot, Sylvie Gimenez, Sandra Nhim, Rima Naït-Saïdi, Marie Frayssinet, Guillaume Dumont, Jean-Paul Boudon, Marin Vabre, Stéphanie Rialle, Rachid Koual, Gael J. Kergoat, Rodney N. Nagoshi, Robert L. Meagher, Emm... | <p>Spodoptera frugiperda, the fall armyworm (FAW), is an important agricultural pest in the Americas and an emerging pest in sub-Saharan Africa, India, East-Asia and Australia, causing damage to major crops such as corn, sorghum and soybean. While... |  | Adaptation, Evolutionary Ecology, Expression Studies, Life History, Speciation | Astrid Groot | | 2018-05-09 13:04:34 | View |
Simulating bacterial evolution forward-in-time
Recommended by Frederic Bertels based on reviews by 3 anonymous reviewers
Jean Cury and colleagues (2021) have developed a protocol to simulate bacterial evolution in SLiM. In contrast to existing methods that depend on the coalescent, SLiM simulates evolution forward in time. SLiM has, up to now, mostly been used to simulate the evolution of eukaryotes (Haller and Messer 2019), but has been adapted here to simulate evolution in bacteria. Forward-in-time simulations are usually computationally very costly. To circumvent this issue, bacterial population sizes are scaled down. One would now expect results to become inaccurate, however, Cury et al. show that scaled-down forwards simulations provide very accurate results (similar to those provided by coalescent simulators) that are consistent with theoretical expectations. Simulations were analyzed and compared to existing methods in simple and slightly more complex scenarios where recombination affects evolution. In all scenarios, simulation results from coalescent methods (fastSimBac (De Maio and Wilson 2017), ms (Hudson 2002)) and scaled-down forwards simulations were very similar, which is very good news indeed.
A biologist not aware of the complexities of forwards, backwards simulations and the coalescent, might now naïvely ask why another simulation method is needed if existing methods perform just as well. To address this question the manuscript closes with a very neat example of what exactly is possible with forwards simulations that cannot be achieved using existing methods. The situation modeled is the growth and evolution of a set of 50 bacteria that are randomly distributed on a petri dish. One side of the petri dish is covered in an antibiotic the other is antibiotic-free. Over time, the bacteria grow and acquire antibiotic resistance mutations until the entire artificial petri dish is covered with a bacterial lawn. This simulation demonstrates that it is possible to simulate extremely complex (e.g. real world) scenarios to, for example, assess whether certain phenomena are expected with our current understanding of bacterial evolution, or whether there are additional forces that need to be taken into account. Hence, forwards simulators could significantly help us to understand what current models can and cannot explain in evolutionary biology.
References
Cury J, Haller BC, Achaz G, Jay F (2021) Simulation of bacterial populations with SLiM. bioRxiv, 2020.09.28.316869, version 5 peer-reviewed and recommended by Peer community in Evolutionary Biology. https://doi.org/10.1101/2020.09.28.316869
De Maio N, Wilson DJ (2017) The Bacterial Sequential Markov Coalescent. Genetics, 206, 333–343. https://doi.org/10.1534/genetics.116.198796
Haller BC, Messer PW (2019) SLiM 3: Forward Genetic Simulations Beyond the Wright–Fisher Model. Molecular Biology and Evolution, 36, 632–637. https://doi.org/10.1093/molbev/msy228
Hudson RR (2002) Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics, 18, 337–338. https://doi.org/10.1093/bioinformatics/18.2.337
| Simulation of bacterial populations with SLiM | Jean Cury, Benjamin C. Haller, Guillaume Achaz, and Flora Jay | <p>Simulation of genomic data is a key tool in population genetics, yet, to date, there is no forward-in-time simulator of bacterial populations that is both computationally efficient and adaptable to a wide range of scenarios. Here we demonstrate... | 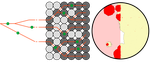 | Bioinformatics & Computational Biology, Population Genetics / Genomics | Frederic Bertels | | 2020-10-02 19:03:42 | View |
Sexual selection goes dynamic
Recommended by Michael D Greenfield based on reviews by Frédéric Guillaume and 1 anonymous reviewer
150 years after Darwin published ‘Descent of man and selection in relation to sex’ (Darwin, 1871), the evolutionary mechanism that he laid out in his treatise continues to fascinate us. Sexual selection is responsible for some of the most spectacular traits among animals, and plants, and it appeals to our interest in all things reproductive and sexual (Bell, 1982). In addition, sexual selection poses some of the more intractable problems in evolutionary biology: Its realm encompasses traits that are subject to markedly different selection pressures, particularly when distinct, yet associated, traits tend to be associated with males, e.g. courtship signals, and with females, e.g. preferences (cf. Ah-King & Ahnesjo, 2013). While separate, such traits cannot evolve independently of each other (Arnqvist & Rowe, 2005), and complex feedback loops and correlations between them are predicted (Greenfield et al., 2014). Traditionally, sexual selection has been modelled under simplifying assumptions, and quantitative genetic approaches that avoided evolutionary dynamics have prevailed. New computing methods may be able to free the field from these constraints, and a trio of theoreticians (Chevalier, De Coligny & Labonne 2020) describe here a novel application of a ‘demo-genetic agent (or individual) based model’, a mouthful hereafter termed DG-ABM, for arriving at a holistic picture of the sexual selection trajectory. The application is built on the premise that traits, e.g. courtship, preference, gamete investment, competitiveness for mates, can influence the genetic architecture, e.g. correlations, of those traits. In turn, the genetic architecture can influence the expression and evolvability of the traits. Much of this influence occurs via demographic features, i.e. social environment, generated by behavioral interactions during sexual advertisement, courtship, mate guarding, parental care, post-mating dispersal, etc.
The authors provide a lengthy verbal description of their model, specifying the genomic and behavioral parameters that can be set and how a ‘run’ may be initialized. There is a link to an internet site where users can then enter their own parameter values and begin exploring hypotheses. Back in the article several simulations illustrate simple tests; e.g. how gamete investment and preference jointly evolve given certain survival costs. One obvious test would have been the preference – courtship genetic correlation that represents the core of Fisherian runaway selection, and it is regrettable that it was not examined under a range of demographic parameters. As presented the author’s DG-ABM appears particularly geared toward mating systems in ‘higher’ vertebrates, where couples form during a discrete mating season and are responsible for most reproduction. It is not clear how applicable the model could be to a full range of mating systems and nuances, including those in arthropods and other invertebrates as well as plants.
What is the likely value of the DG-ABM for sexual selection researchers? We will not be able to evaluate its potential impact until readers with specialized understanding of a question and taxon begin exploring and comparing their results with prior expectations. Of course, lack of congruence with earlier predictions would not invalidate the model. Hopefully, some of these specialists will have opportunities for comparing results with pertinent empirical data.
References
Ah-King, M. and Ahnesjo, I. 2013. The ‘sex role’ concept: An overview and evaluation Evolutionary Biology, 40, 461-470. doi: https://doi.org/10.1007/s11692-013-9226-7
Arnqvist, G. and Rowe, L. 2005. Sexual Conflict. Princeton University Press, Princeton. doi: https://doi.org/10.1515/9781400850600
Bell, G. 1982. The Masterpiece of Nature: The Evolution and Genetics of Sexuality. University of California Press, Berkeley.
Chevalier, L., De Coligny, F. and Labonne, J. (2020) A demogenetic individual based model for the evolution of traits and genome architecture under sexual selection. bioRxiv, 2020.04.01.014514, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: https://doi.org/10.1101/2020.04.01.014514
Darwin, C. 1871. The Descent of Man and Selection in Relation to Sex. J. Murray, London.
Greenfield, M.D., Alem, S., Limousin, D. and Bailey, N.W. 2014. The dilemma of Fisherian sexual selection: Mate choice for indirect benefits despite rarity and overall weakness of trait-preference genetic correlation. Evolution, 68, 3524-3536. doi: https://doi.org/10.1111/evo.12542
| A demogenetic agent based model for the evolution of traits and genome architecture under sexual selection | Louise Chevalier, François de Coligny, Jacques Labonne | <p>Sexual selection has long been known to favor the evolution of mating behaviors such as mate preference and competitiveness, and to affect their genetic architecture, for instance by favoring genetic correlation between some traits. Reciprocall... | 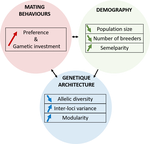 | Adaptation, Behavior & Social Evolution, Evolutionary Dynamics, Evolutionary Theory, Life History, Population Genetics / Genomics, Sexual Selection | Michael D Greenfield | | 2020-04-02 14:44:25 | View |
Genotypic sex shapes maternal care in the African Pygmy mouse, Mus minutoides
Louise D Heitzmann, Marie Challe, Julie Perez, Laia Castell, Evelyne Galibert, Agnes Martin, Emmanuel Valjent, Frederic Veyrunes
https://doi.org/10.1101/2022.04.05.487174
Effect of sex chromosomes on mammalian behaviour: a case study in pygmy mice
Recommended by Gabriel Marais and Trine Bilde based on reviews by Marion Anne-Lise Picard, Caroline Hu and 1 anonymous reviewer based on reviews by Marion Anne-Lise Picard, Caroline Hu and 1 anonymous reviewer
In mammals, it is well documented that sexual dimorphism and in particular sex differences in behaviour are fine-tuned by gonadal hormonal profiles. For example, in lemurs, where female social dominance is common, the level of testosterone in females is unusually high compared to that of other primate females (Petty and Drea 2015).
Recent studies however suggest that gonadal hormones might not be the only biological factor involved in establishing sexual dimorphism, sex chromosomes might also play a role. The four core genotype (FCG) model and other similar systems allowing to decouple phenotypic and genotypic sex in mice have provided very convincing evidence of such a role (Gatewood et al. 2006; Arnold and Chen 2009; Arnold 2020a, 2020b). This however is a new field of research and the role of sex chromosomes in establishing sexually dimorphic behaviours has not been studied very much yet. Moreover, the FCG model has some limits. Sry, the male determinant gene on the mammalian Y chromosome might be involved in some sex differences in neuroanatomy, but Sry is always associated with maleness in the FCG model, and this potential role of Sry cannot be studied using this system.
Heitzmann et al. have used a natural system to approach these questions. They worked on the African Pygmy mouse, Mus minutoides, in which a modified X chromosome called X* can feminize X*Y individuals, which offers a great opportunity for elegant experiments on the effects of sex chromosomes versus hormones on behaviour. They focused on maternal care and compared pup retrieval, nest quality, and mother-pup interactions in XX, X*X and X*Y females. They found that X*Y females are significantly better at retrieving pups than other females. They are also much more aggressive towards the fathers than other females, preventing paternal care. They build nests of poorer quality but have similar interactions with pups compared to other females. Importantly, no significant differences were found between XX and X*X females for these traits, which points to an effect of the Y chromosome in explaining the differences between X*Y and other females (XX, X*X). Also, another work from the same group showed similar gonadal hormone levels in all the females (Veyrunes et al. 2022).
Heitzmann et al. made a number of predictions based on what is known about the neuroanatomy of rodents which might explain such behaviours. Using cytology, they looked for differences in neuron numbers in the hypothalamus involved in the oxytocin, vasopressin and dopaminergic pathways in XX, X*X and X*Y females, but could not find any significant effects. However, this part of their work relied on very small sample sizes and they used virgin females instead of mothers for ethical reasons, which greatly limited the analysis.
Interestingly, X*Y females have a higher reproductive performance than XX and X*X ones, which compensate for the cost of producing unviable YY embryos and certainly contribute to maintaining a high frequency of X* in many African pygmy mice populations (Saunders et al. 2014, 2022). X*Y females are probably solitary mothers contrary to other females, and Heitzmann et al. have uncovered a divergent female strategy in this species. Their work points out the role of sex chromosomes in establishing sex differences in behaviours.
References
Arnold AP (2020a) Sexual differentiation of brain and other tissues: Five questions for the next 50 years. Hormones and Behavior, 120, 104691. https://doi.org/10.1016/j.yhbeh.2020.104691
Arnold AP (2020b) Four Core Genotypes and XY* mouse models: Update on impact on SABV research. Neuroscience & Biobehavioral Reviews, 119, 1–8. https://doi.org/10.1016/j.neubiorev.2020.09.021
Arnold AP, Chen X (2009) What does the “four core genotypes” mouse model tell us about sex differences in the brain and other tissues? Frontiers in Neuroendocrinology, 30, 1–9. https://doi.org/10.1016/j.yfrne.2008.11.001
Gatewood JD, Wills A, Shetty S, Xu J, Arnold AP, Burgoyne PS, Rissman EF (2006) Sex Chromosome Complement and Gonadal Sex Influence Aggressive and Parental Behaviors in Mice. Journal of Neuroscience, 26, 2335–2342. https://doi.org/10.1523/JNEUROSCI.3743-05.2006
Heitzmann LD, Challe M, Perez J, Castell L, Galibert E, Martin A, Valjent E, Veyrunes F (2022) Genotypic sex shapes maternal care in the African Pygmy mouse, Mus minutoides. bioRxiv, 2022.04.05.487174, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.04.05.487174
Petty JMA, Drea CM (2015) Female rule in lemurs is ancestral and hormonally mediated. Scientific Reports, 5, 9631. https://doi.org/10.1038/srep09631
Saunders PA, Perez J, Rahmoun M, Ronce O, Crochet P-A, Veyrunes F (2014) Xy Females Do Better Than the Xx in the African Pygmy Mouse, Mus Minutoides. Evolution, 68, 2119–2127. https://doi.org/10.1111/evo.12387
Saunders PA, Perez J, Ronce O, Veyrunes F (2022) Multiple sex chromosome drivers in a mammal with three sex chromosomes. Current Biology, 32, 2001-2010.e3. https://doi.org/10.1016/j.cub.2022.03.029
Veyrunes F, Perez J, Heitzmann L, Saunders PA, Givalois L (2022) Separating the effects of sex hormones and sex chromosomes on behavior in the African pygmy mouse Mus minutoides, a species with XY female sex reversal. bioRxiv, 2022.07.11.499546. https://doi.org/10.1101/2022.07.11.499546
| Genotypic sex shapes maternal care in the African Pygmy mouse, Mus minutoides | Louise D Heitzmann, Marie Challe, Julie Perez, Laia Castell, Evelyne Galibert, Agnes Martin, Emmanuel Valjent, Frederic Veyrunes | <p>Sexually dimorphic behaviours, such as parental care, have long been thought to be driven mostly, if not exclusively, by gonadal hormones. In the past two decades, a few studies have challenged this view, highlighting the direct influence of th... | 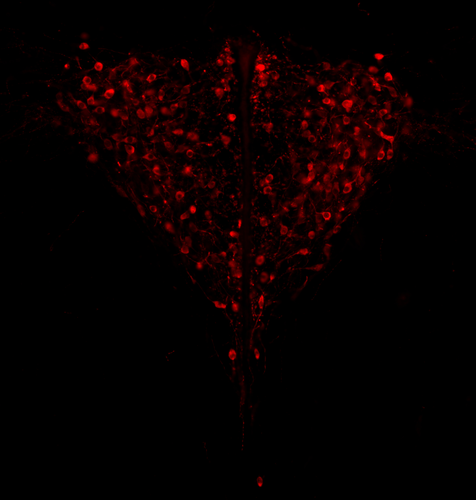 | Behavior & Social Evolution, Evolutionary Ecology, Reproduction and Sex | Gabriel Marais | | 2022-04-08 20:09:58 | View |
Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates
Rylan Shearn, Alison E. Wright, Sylvain Mousset, Corinne Régis, Simon Penel, Jean-François Lemaitre, Guillaume Douay, Brigitte Crouau-Roy, Emilie Lecompte, Gabriel A.B. Marais
https://doi.org/10.1101/445072
Studying genetic antagonisms as drivers of genome evolution
Recommended by Mathieu Joron based on reviews by Qi Zhou and 3 anonymous reviewers
Sex chromosomes are special in the genome because they are often highly differentiated over much of their lengths and marked by degenerative evolution of their gene content. Understanding why sex chromosomes differentiate requires deciphering the forces driving their recombination patterns. Suppression of recombination may be subject to selection, notably because of functional effects of locking together variation at different traits, as well as longer-term consequences of the inefficient purge of deleterious mutations, both of which may contribute to patterns of differentiation [1]. As an example, male and female functions may reveal intrinsic antagonisms over the optimal genotypes at certain genes or certain combinations of interacting genes. As a result, selection may favour the recruitment of rearrangements blocking recombination and maintaining the association of sex-antagonistic allele combinations with the sex-determining locus.
The hypothesis that sexually antagonistic selection might drive recombination suppression along the sex chromosomes is not new, but there are surprisingly few studies examining this empirically [1]. Support mainly comes from the study of guppy populations Poecilia reticulata in which the level of sexual dimorphism (notably due to male ornaments, subject to sexual selection) varies among populations, and was found to correlate with the length of the non-recombining region on the sex chromosome [2]. But the link is not always that clear. For instance in the fungus Microbotryum violaceum, the mating type loci is characterized by adjacent segments with recombination suppression, despite the near absence of functional differentiation between mating types [3].
In this study, Shearn and colleagues [4] explore the patterns of recombination suppression on the sex chromosomes of primates. X and Y chromosomes are strongly differentiated, except in a small region where they recombine with each other, the pseudoautosomal region (PAR). In the clade of apes and monkeys, including humans, large rearrangements have extended the non recombining region stepwise, eroding the PAR. Could this be driven by sexually antagonistic selection in a clade showing strong sexual differentiation?
To evaluate this idea, Shearn et al. have compared the structure of recombination in apes and monkeys to their sister clade with lower levels of sexual dimorphism, the lemurs and the lorises. If sexual antagonism was important in shaping recombination suppression, and assuming lower measures of sexual dimorphism reflect lower sexual antagonism [5], then lemurs and lorises would be predicted to show a shorter non-recombining region than apes and monkeys.
Lemurs and lorises were terra incognita in terms of genomic research on the sex chromosomes, so Shearn et al. have sequenced the genomes of males and females of different species. To assess whether sequences came from a recombining or non-recombining segment, they used coverage information in males vs females to identify sequences on the X whose copy on the Y is absent or too divergent to map, indicating long-term differentiation (absence of recombination). This approach reveals that the two lineages have undergone different recombination dynamics since they split from their common ancestor: regions which have undergone further structural rearrangements extending the non-recombining region in apes and monkeys, have continued to recombine normally in lemurs and lorises. Consistent with the prediction, macroevolutionary variation in the differentiation of males and females is indeed accompanied by variation in the size of the non-recombining region on the sex chromosome.
Sex chromosomes are excellent examples of how genomes are shaped by selection. By directly exploring recombination patterns on the sex chromosome across all extant primate groups, this study comes as a nice addition to the short series of empirical studies evaluating whether sexual antagonism may drive certain aspects of genome structure. The sexual selection causing sometimes spectacular morphological or behavioural differences between sexes in many animals may be the visible tip of the iceberg of all the antagonisms that characterise male vs. female functions generally [5]. Further research should bring insight into how different flavours or intensities of antagonistic selection can contribute to shape genome variation.
References
[1] Charlesworth D (2017) Evolution of recombination rates between sex chromosomes. Philosophical Transactions of the Royal Society B: Biological Sciences, 372, 20160456. https://doi.org/10.1098/rstb.2016.0456
[2] Wright AE, Darolti I, Bloch NI, Oostra V, Sandkam B, Buechel SD, Kolm N, Breden F, Vicoso B, Mank JE (2017) Convergent recombination suppression suggests role of sexual selection in guppy sex chromosome formation. Nature Communications, 8, 14251. https://doi.org/10.1038/ncomms14251
[3] Branco S, Badouin H, Vega RCR de la, Gouzy J, Carpentier F, Aguileta G, Siguenza S, Brandenburg J-T, Coelho MA, Hood ME, Giraud T (2017) Evolutionary strata on young mating-type chromosomes despite the lack of sexual antagonism. Proceedings of the National Academy of Sciences, 114, 7067–7072. https://doi.org/10.1073/pnas.1701658114
[4] Shearn R, Wright AE, Mousset S, Régis C, Penel S, Lemaitre J-F, Douay G, Crouau-Roy B, Lecompte E, Marais GAB (2020) Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates. bioRxiv, 445072. https://doi.org/10.1101/445072
[5] Connallon T, Clark AG (2014) Evolutionary inevitability of sexual antagonism. Proceedings of the Royal Society B: Biological Sciences, 281, 20132123. https://doi.org/10.1098/rspb.2013.2123
| Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates | Rylan Shearn, Alison E. Wright, Sylvain Mousset, Corinne Régis, Simon Penel, Jean-François Lemaitre, Guillaume Douay, Brigitte Crouau-Roy, Emilie Lecompte, Gabriel A.B. Marais | <p>Sex chromosomes are typically comprised of a non-recombining region and a recombining pseudoautosomal region. Accurately quantifying the relative size of these regions is critical for sex chromosome biology both from a functional (i.e. number o... | 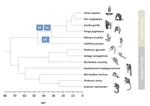 | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Reproduction and Sex, Sexual Selection | Mathieu Joron | | 2019-02-04 15:16:32 | View |
Variation in competitive ability with mating system, ploidy and range expansion in four Capsella species
Xuyue Yang, Martin Lascoux and Sylvain Glémin
https://doi.org/10.1101/214866
When ecology meets genetics: Towards an integrated understanding of mating system transitions and diversity
Recommended by Sylvain Billiard and Henrique Teotonio based on reviews by Yaniv Brandvain, Henrique Teotonio and 1 anonymous reviewer
In the 19th century, C. Darwin and F. Delpino engaged in a debate about the success of species with different reproduction modes, with the later favouring the idea that monoecious plants capable of autonomous selfing could spread more easily than dioecious plants (or self-incompatible hermaphroditic plants) if cross-pollination opportunities were limited [1]. Since then, debate has never faded about how natural selection is responsible for transitions to selfing and can explain the diversity and distribution of reproduction modes we observe in the natural world [2, 3].
Explanations for mating systems diversity, and transitions to selfing in particular, generally fall into two categories: either genetic or ecological. On the genetic side, many theoretical works showed a critical role for mutation load and inbreeding depression, transmission advantage and reproductive assurance in the evolution of selfing, e.g. [4]. Many experimental works were conducted to test theoretical hypotheses and predictions, especially regarding the magnitude of inbreeding depression; see [5] for a review. Ecologically, the presence of selfing populations is usually correlated with fragmented and harsh habitats, on the periphery of ancestral outcrossing populations. The cause of this distribution could be that selfers are better dispersers and colonizers than outcrossers, or variations in other life-history traits [6]. Yet, few experiments were run to assess whether selfing species or populations have effectively different ecological characteristics, and even scarcer are experiments evaluating both the roles of mutational load and life-history traits evolution. This is the aim of the present study by X. Yang et al [7].
The study of Yang et al [7], together with that of Petrone Mendoza et al. [8], supervised by S. Glémin and M. Lascoux, is probably one of the first to conduct experiments where the competitive abilities are compared between and within species. Using 4 species of the Capsella genus, annual plants from the mustard family, they tested the theoretical predictions that i) the transition from outcrossing to selfing resulted in reduced competitive ability at higher densities, because of the accumulation of deleterious mutations and/or the evolution of life-history traits in an open habitat and a colonization/dispersal trade-off; ii) that reduced competitive ability of selfers should be less pronounced in polyploid then diploid species because the effect of partially recessive deleterious mutations would be buffered; and iii) that competitive ability of selfers should decline with historical range expansion because of the expansion load [9].
Of the 4 Capsella species studied, only one of them, presumably the ancestral, is a diploid outcrosser with a small distribution but large population sizes. The three other species are selfers, two diploids with independent histories of transitions from outcrossing, and another, tetraploid, resulting from a recent hybridization between one of the diploid selfer and the diploid outcrossing ancestor. Many accessions from each species were sampled and individuals assayed for their competitive ability against a tester species or alone, for vegetative and reproductive traits. The measured vegetative traits (rosette surface at two stages, growth rate and flowering probability) showed no differentiation between selfers and outcrossers. To the contrary, reproductive traits (number of flowers) followed theoretical predictions: selfing species are more sensitive to competition than the outcrossing species, with polyploid selfing species being intermediate between the diploid selfers and the diploid outcrosser, and within the tetraploid selfing species (where sampling was quite significant across a large geographical range) sensitivity to competition increased with range expansion.
The study of Yang et al. [7] suffers from several limitations, such that alternative explanations cannot be discarded in the absence of further experimental data. They nonetheless provide the reader with a nice discussion and prospects on how to untwine the causes and the consequences of transitions to selfing. Their study also brings up to date questions about the joint evolution of mating system and life-history traits, which needs a renewed interest from an empirical and theoretical point of view. The results of Yang et al. raise for instance the question of whether it is indeed expected that only reproductive traits, and not vegetative traits, should evolve with the transition to selfing.
The recommandation and evaluation of this paper have been made in collaboration with Thomas Lesaffre.
References
[1] Darwin, C. R. (1876). The effects of cross and self fertilization in the vegetable kingdom. London: Murray.
[2] Stebbins, G. L. (1957). Self fertilization and population variability in the higher plants. The American Naturalist, 91, 337-354. doi: 10.1086/281999
[3] Harder, L.D. & Barrett, S. C. H. (2006). Ecology and evolution of flowers. Oxford: Oxford University Press.
[4] Porcher, E. & Lande, R. (2005). The evolution of self-fertilization and inbreeding depression under pollen discounting and pollen limitation. Journal of Evolutionary Biology, 18(3), 497-508. doi: 10.1111/j.1420-9101.2005.00905.x
[5] Winn, A.A., et al. (2011). Analysis of inbreeding depression in mixed-mating plants provides evidence for selective interference and stable mixed mating. Evolution, 65(12), 3339-3359. doi: 10.1111/j.1558-5646.2011.01462.x
[6] Munoz, F., Violle, C. & Cheptou, P.-O. (2016). CSR ecological strategies and plant mating systems: outcrossing increases with competitiveness but stress-tolerance is related to mixed mating. Oikos, 125(9), 1296-1303. doi: 10.1111/oik.02328
[7] Yang, X., Lascoux, M. & Glémin, S (2018). Variation in competitive ability with mating system, ploidy and range expansion in four Capsella species. bioRxiv, 214866, ver. 5 recommended and peer-reviewed by PCI Evol Biol. doi: 10.1101/214866
[8] Petrone Mendoza, S., Lascoux, M. & Glémin, S. (2018). Competitive ability of Capsella species with different mating systems and ploidy levels. Annals of Botany 121(6), 1257-1264. doi: 10.1093/aob/mcy014
[9] Peischl, S. & Excoffier, L. (2015). Expansion load: recessive mutations and the role of standing genetic variation. Molecular Ecology, 24(9): 2084-2094. doi: 10.1111/mec.13154
| Variation in competitive ability with mating system, ploidy and range expansion in four Capsella species | Xuyue Yang, Martin Lascoux and Sylvain Glémin | <p>Self-fertilization is often associated with ecological traits corresponding to the ruderal strategy in Grime’s Competitive-Stress-tolerant-Ruderal (CSR) classification of ecological strategies. Consequently, selfers are expected to be less comp... |  | Evolutionary Ecology, Population Genetics / Genomics, Reproduction and Sex, Species interactions | Sylvain Billiard | | 2017-11-06 19:54:52 | View |
New insights into the population genetics of partially clonal organisms: when seagrass data meet theoretical expectations
Arnaud-Haond, Sophie, Stoeckel, Solenn, and Bailleul, Diane
https://arxiv.org/abs/1902.10240v6
Inferring rates of clonal versus sexual reproduction from population genetics data
Recommended by Olivier J Hardy based on reviews by Ludwig TRIEST, Stacy Krueger-Hadfield and 1 anonymous reviewer
In partially clonal organisms, genetic markers are often used to characterize the genotypic diversity of populations and infer thereof the relative importance of clonal versus sexual reproduction. Most studies report a measure of genotypic diversity based on a ratio, R, of the number of distinct multilocus genotypes over the sample size, and qualitatively interpret high / low R as indicating the prevalence of sexual / clonal reproduction. However, a theoretical framework allowing to quantify the relative rates of clonal versus sexual reproduction from genotypic diversity is still lacking, except using temporal sampling. Moreover, R is intrinsically highly dependent on sample size and sample design, while alternative measures of genotypic diversity are more robust to sample size, like D*, which is equivalent to the Gini-Simpson diversity index applied to multilocus genotypes. Another potential indicator of reproductive strategies is the inbreeding coefficient, Fis, because population genetics theory predicts that clonal reproduction should lead to negative Fis, at least when the sexual reproduction component occurs through random mating. Taking advantage of this prediction, Arnaud-Haond et al. [1] reanalysed genetic data from 165 populations of four partially clonal seagrass species sampled in a standardized way. They found positive correlations between Fis and both R and D* within each species, reflecting variation in the relative rates of sexual versus clonal reproduction among populations. Moreover, the differences of mean genotypic diversity and Fis values among species were also consistent with their known differences in reproductive strategies. Arnaud-Haond et al. [1] also conclude that previous works based on the interpretation of R generally lead to underestimate the prevalence of clonality in seagrasses. Arnaud-Haond et al. [1] confirm experimentally that Fis merits to be interpreted more properly than usually done when inferring rates of clonal reproduction from population genetics data of species reproducing both sexually and clonally. An advantage of Fis is that it is much less affected by sample size than R, and thus should be more reliable when comparing studies differing in sample design. Hence, when the rate of clonal reproduction becomes significant, we expect Fis < 0 and D* < 1. I expect these two indicators of clonality to be complementary because they rely on different consequences of clonality on pattern of genetic variation. Nevertheless, both measures can be affected by other factors. For example, null alleles, selfing or biparental inbreeding can pull Fis upwards, potentially eliminating the signature of clonal reproduction. Similarly, D* (and other measures of genotypic diversity) can be low because the polymorphism of the genetic markers used is too limited or because sexual reproduction often occurs through selfing, eventually resulting in highly similar homozygous genotypes.
The work of Arnaud-Haond et al. [1] shows that the populations genetics of partially clonal organisms should be better studied, an endeavour encompassed in a companion paper using numerical simulations [2]. A further step that remains to be accomplished is to build a mathematical framework for developing estimators of rates of clonal versus sexual reproduction based on genotypic diversity.
References
[1] Arnaud-Haond, S., Stoeckel, S., and Bailleul, D. (2019). New insights into the population genetics of partially clonal organisms: when seagrass data meet theoretical expectations. ArXiv:1902.10240 [q-Bio], v6 peer-reviewed and recommended by Peer Community in Evolutionary Biology. Retrieved from http://arxiv.org/abs/1902.10240
[2] Stoeckel, S., Porro, B., and Arnaud-Haond, S. (2019). The discernible and hidden effects of clonality on the genotypic and genetic states of populations: improving our estimation of clonal rates. ArXiv:1902.09365 [q-Bio], v4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. Retrieved from http://arxiv.org/abs/1902.09365
| New insights into the population genetics of partially clonal organisms: when seagrass data meet theoretical expectations | Arnaud-Haond, Sophie, Stoeckel, Solenn, and Bailleul, Diane | <p>Seagrass meadows are among the most important coastal ecosystems, in terms of both spatial extent and ecosystem services, but they are also declining worldwide. Understanding the drivers of seagrass meadow dynamics is essential for designing so... |  | Evolutionary Ecology, Population Genetics / Genomics, Reproduction and Sex | Olivier J Hardy | | 2019-03-01 21:57:34 | View |
Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands
Christelle Fraïsse, Anne Haguenauer, Karin Gerard, Alexandra Anh-Thu Weber, Nicolas Bierne, Anne Chenuil
https://doi.org/10.1101/239244
Introgression from related species reveals fine-scale structure in an isolated population of mussels and causes patterns of genetic-environment associations
Recommended by Marianne Elias based on reviews by Thomas Broquet and Tatiana Giraud
Assessing population connectivity is central to understanding population dynamics, and is therefore of great importance in evolutionary biology and conservation biology. In the marine realm, the apparent absence of physical barriers, large population sizes and high dispersal capacities of most organisms often result in no detectable structure, thereby hindering inferences of population connectivity. In a review paper, Gagnaire et al. [1] propose several ideas to improve detection of population connectivity. Notably, using simulations they show that under certain circumstances introgression from one species into another may reveal cryptic population structure within that second species.
The isolated Kerguelen archipelago in the south of Indian Ocean represents a typical situation where the structure of coastal marine organisms is expected to be difficult to detect. In an elegant genomic study, Fraïsse et al. [2] take advantage of introgression from foreign lineages to infer fine-grained population structure in a population of mussels around the Kerguelen archipelago, and investigate its association with environmental variables. Using a large panel of genome-wide markers (GBS) and applying a range of methods that unravel patterns of divergence and gene flow among lineages, they first find that the Kerguelen population is highly admixed, with a major genetic background corresponding to the southern mussel lineage Mytilus platensis introgressed by three northern lineages. By selecting a panel of loci enriched in ancestry-informative SNPs (ie, SNPs highly differentiated among northern lineages) they then detect a fine-scale genetic structure around the Kerguelen archipelago, and identify a major connectivity break. They further show an associating between the genetic structure and environmental variables, particularly the presence of Macrocystis kelp, a marker of habitat exposure to waves (a feature repeatedly evidenced to be important for mussels). While such association pattern could lead to the interpretation that differentiated SNPs correspond to loci directly under selection or linked with such loci, and even be considered as support for adaptive introgression, Fraïsse et al. [2] convincingly show by performing simulations that the genetic-environment association detected can be entirely explained by dispersal barriers associated with environmental variables (habitat-associated connectivity). They also explain why the association is better detected by ancestry-informative SNPs as predicted by Gagnaire et al. [1]. In addition, intrinsic genetic incompatibilities, which reduce gene flow, tend to become trapped at ecotones due to ecological selection, even when loci causing genetic incompatibilities are unlinked with loci involved in adaption to local ecological conditions (Bierne et al. [3]’s coupling hypothesis), leading to correlations between environmental variables and loci not involved in local adaptation. Notably, in Fraïsse et al. [2]’s study, the association between the kelp and ancestry-informative alleles is not consistent throughout the archipelago, casting further doubt on the implication of these alleles in local adaptation.
The study of Fraïsse et al. [2] is therefore an important contribution to evolutionary biology because 1) it provides an empirical demonstration that alleles of foreign origin can be pivotal to detect fine-scale connectivity patterns and 2) it represents a test case of Bierne et al. [3]’s coupling hypothesis, whereby introgressed alleles also enhance patterns of genetic-environment associations. Since genomic scan or GWAS approaches fail to clearly reveal loci involved in local adaptation, how can we disentangle environment-driven selection from intrinsic reproductive barriers and habitat-associated connectivity? A related question is whether we can reliably identify cases of adaptive introgression, which have increasingly been put forward as a mechanism involved in adaptation [4]. Unfortunately, there is no easy answer, and the safest way to go is to rely – where possible – on independent information [5], in particular functional studies of the detected loci, as is for example the case in the mimetic butterfly Heliconius literature (e. g., [6]) where several loci controlling colour pattern variation are well characterized.
References
[1] Gagnaire, P.-A., Broquet, T., Aurelle, D., Viard, F., Souissi, A., Bonhomme, F., Arnaud-Haond, S., & Bierne, N. (2015). Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evolutionary Applications, 8, 769–786. doi: 10.1111/eva.12288
[2] Fraïsse, C., Haguenauer, A., Gerard, K., Weber, A. A.-T., Bierne, N., & Chenuil, A. (2018). Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands. bioRxiv, 239244, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/239244
[3] Bierne, N., Welch, J., Loire, E., Bonhomme, F., & David, P. (2011). The coupling hypothesis: why genome scans may fail to map local adaptation genes. Molecular Ecology, 20, 2044–2072. doi: 10.1111/j.1365-294X.2011.05080.x
[4] Hedrick, P. W. (2013). Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Molecular Ecology, 22, 4606–4618. doi: 10.1111/mec.12415
[5] Ravinet, M., Faria, R., Butlin, R. K., Galindo, J., Bierne, N., Rafajlović, M., Noor, M. A. F., Mehlig, B., & Westram, A. M. (2017). Interpreting the genomic landscape of speciation: a road map for finding barriers to gene flow. Journal of Evolutionary Biology, 30, 1450–1477. doi: 10.1111/jeb.13047.
[6] Jay, P., Whibley, A., Frézal, L., Rodríguez de Cara, M. A., Nowell, R. W., Mallet, J., Dasmahapatra, K. K., & Joron, M. (2018). Supergene evolution triggered by the introgression of a chromosomal inversion. Current Biology, 28, 1839–1845.e3. doi: 10.1016/j.cub.2018.04.072
| Fine-grained habitat-associated genetic connectivity in an admixed population of mussels in the small isolated Kerguelen Islands | Christelle Fraïsse, Anne Haguenauer, Karin Gerard, Alexandra Anh-Thu Weber, Nicolas Bierne, Anne Chenuil | <p>Reticulated evolution -i.e. secondary introgression / admixture between sister taxa- is increasingly recognized as playing a key role in structuring infra-specific genetic variation and revealing cryptic genetic connectivity patterns. When admi... |  | Hybridization / Introgression, Phylogeography & Biogeography, Population Genetics / Genomics | Marianne Elias | | 2017-12-28 14:16:16 | View |
Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic data
Emeric Figuet, Marion Ballenghien, Nicolas Lartillot, Nicolas Galtier
https://doi.org/10.1101/139147
Predicting small ancestors using contemporary genomes of large mammals
Recommended by Bruce Rannala based on reviews by Bruce Rannala and 1 anonymous reviewer
Recent methodological developments and increased genome sequencing efforts have introduced the tantalizing possibility of inferring ancestral phenotypes using DNA from contemporary species. One intriguing application of this idea is to exploit the apparent correlation between substitution rates and body size to infer ancestral species' body sizes using the inferred patterns of substitution rate variation among species lineages based on genomes of extant species [1].
The recommended paper by Figuet et al. [2] examines the utility of such approaches by analyzing the Cetartiodactyla, a clade of large mammals that have mostly well resolved phylogenetic relationships and a reasonably good fossil record. This combination of genomic data and fossils allows a direct comparison between body size predictions obtained from the genomic data and empirical evidence from the fossil record. If predictions seem good in groups such as the Cetartiodactyla, where there is independent evidence from the fossil record, this would increase the credibility of predictions made for species with less abundant fossils.
Figuet et al. [2] analyze transcriptome data for 41 species and report a significant effect of body mass on overall substitution rate, synonymous vs. non-synonymous rates, and the dynamics of GC-content, thus allowing a prediction of small ancestral body size in this group despite the fact that the extant species that were analyzed are nearly all large.
A comparative method based solely on morphology and phylogenetic relationships would be very unlikely to make such a prediction. There are many sources of uncertainty in the variables and parameters associated with these types of approaches: phylogenetic uncertainty (topology and branch lengths), uncertainty about inferred substitution rates, and so on. Although the authors do not account for all these sources of uncertainty the fact that their predicted body sizes appear sensible is encouraging and undoubtedly the methods will become more statistically sophisticated over time.
References
[1] Romiguier J, Ranwez V, Douzery EJP and Galtier N. 2013. Genomic evidence for large, long-lived ancestors to placental mammals. Molecular Biology and Evolution 30: 5–13. doi: 10.1093/molbev/mss211
[2] Figuet E, Ballenghien M, Lartillot N and Galtier N. 2017. Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic data. bioRxiv, ver. 3 of 4th December 2017. 139147. doi: 10.1101/139147
| Reconstruction of body mass evolution in the Cetartiodactyla and mammals using phylogenomic data | Emeric Figuet, Marion Ballenghien, Nicolas Lartillot, Nicolas Galtier | <p>Reconstructing ancestral characters on a phylogeny is an arduous task because the observed states at the tips of the tree correspond to a single realization of the underlying evolutionary process. Recently, it was proposed that ancestral traits... |  | Genome Evolution, Life History, Macroevolution, Molecular Evolution, Phylogenetics / Phylogenomics | Bruce Rannala | | 2017-05-18 15:28:58 | View |
Shifts from pulled to pushed range expansions caused by reduction of landscape connectivity
Maxime Dahirel, Aline Bertin, Marjorie Haond, Aurélie Blin, Eric Lombaert, Vincent Calcagno, Simon Fellous, Ludovic Mailleret, Thibaut Malausa, Elodie Vercken
https://doi.org/10.1101/2020.05.13.092775
The push and pull between theory and data in understanding the dynamics of invasion
Recommended by Ben Phillips based on reviews by Laura Naslund and 2 anonymous reviewers
Exciting times are afoot for those of us interested in the ecology and evolution of invasive populations. Recent years have seen evolutionary process woven firmly into our understanding of invasions (Miller et al. 2020). This integration has inspired a welter of empirical and theoretical work. We have moved from field observations and verbal models to replicate experiments and sophisticated mathematical models. Progress has been rapid, and we have seen science at its best; an intimate discussion between theory and data.
An area currently under very active development is our understanding of pushed invasions. Here a population spreads through space driven, not by dispersal and growth originating at the leading tip of the invasion, but by dispersal and growth originating deeper in the bulk of the population. These pushed invasions may be quite common – they result when per capita growth and dispersal rates are higher in the bulk of the wave than at the leading tip. They result from a range of well-known phenomena, including Allee effects and density-dependent dispersal (Gandhi et al. 2016; Bîrzu et al. 2019). Pushed invasions travel faster than we would expect given growth and dispersal rates on the leading tip, and they lose genetic diversity more slowly than classical pulled invasions (Roques et al. 2012; Haond et al. 2018; Bîrzu et al. 2019).
Well… in theory, anyway. The theory on pushed waves has momentarily streaked ahead of the empirical work, because empirical systems for studying pushed invasions are rare (though see Gandhi et al. 2016; Gandhi, Korolev, and Gore 2019). In this paper, Dahirel and colleagues (2020) make the argument that we may be able to generate pushed invasions in laboratory systems simply by reducing the connectedness of our experimental landscapes. If true, we might have a simple tool for turning many of our established experimental systems into systems for studying pushed dynamics.
It’s a nice idea, and the paper goes to careful lengths to explore the possibility in their lab system (a parasitoid wasp, Trichogramma). They run experiments on replicate wasp populations comparing strongly- v poorly-connected arrays, and estimate the resulting invasion speeds and rate of diversity loss. They also build a simulation model of the system, allowing them to explore in-silico a range of possible processes underlying their results.
As well as developing these parallel systems, Dahirel and colleagues (2020) go to careful lengths to develop statistical analyses that allow inference on key parameters, and they apply these analyses to both the experimental and simulation data. They have been motivated to apply methods that might be used in both laboratory and field settings to help classify invasions.
Ultimately, they found reasonable evidence that their poorly-connected habitat did induce a pushed dynamic. Their poorly connected invasions travelled faster than they should have if they were pulled, they lost diversity more slowly than the highly connected habitat, and replicates with a higher carrying capacity tended to have higher invasion speeds. All in line with expectations of a pushed dynamic. Interestingly, however, their simulation results suggest that they probably got this perfect result for unexpected reasons. The strong hint is that their poorly-connected habitat induced density dependent dispersal in the wasps. Without this effect, their simulations suggest they should have seen diversity decreasing much more rapidly than it did.
There is a nuanced, thoughtful, and carefully argued discussion about all this in the paper, and it is worth reading. There is much of value in this paper. Theirs is not a perfect empirical system in which all the model assumptions are met and in which huge population sizes make stochastic effects negligible. Here is a system one step closer to the messy reality of biology. The struggle to align this system with new theory has been worth the effort. Not only does it give us hope that we might usefully be able to discriminate between classes of invasions using real-world data, but it hints at a rule that Tolstoy might have expressed this way: all pulled invasions are alike, each pushed invasion is pushed in its own way.
References
Bîrzu, G., Matin, S., Hallatschek, O., and Korolev, K. S. (2019). Genetic drift in range expansions is very sensitive to density dependence in dispersal and growth. Ecology Letters, 22(11), 1817-1827. doi: https://doi.org/10.1111/ele.13364
Dahirel, M., Bertin, A., Haond, M., Blin, A., Lombaert, E., Calcagno, V., Fellous, S., Mailleret, L., Malausa, T., and Vercken, E. (2020). Shifts from pulled to pushed range expansions caused by reduction of landscape connectivity. bioRxiv, 2020.05.13.092775, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. https://doi.org/10.1101/2020.05.13.092775
Gandhi, S. R., Korolev, K. S., and Gore, J. (2019). Cooperation mitigates diversity loss in a spatially expanding microbial population. Proceedings of the National Academy of Sciences, 116(47), 23582-23587. doi: https://doi.org/10.1073/pnas.1910075116
Gandhi, S. R., Yurtsev, E. A., Korolev, K. S., and Gore, J. (2016). Range expansions transition from pulled to pushed waves as growth becomes more cooperative in an experimental microbial population. Proceedings of the National Academy of Sciences, 113(25), 6922-6927. doi: https://doi.org/10.1073/pnas.1521056113
Haond, M., Morel-Journel, T., Lombaert, E., Vercken, E., Mailleret, L. and Roques, L. (2018). When higher carrying capacities lead to faster propagation (2018), bioRxiv, 307322, ver. 4 peer-reviewed and recommended by Peer Community in Ecology. https://doi.org/10.1101/307322
Miller et al. (2020). Eco‐evolutionary dynamics of range expansion. Ecology, 101(10), e03139. doi: https://doi.org/10.1002/ecy.3139
Roques, L., Garnier, J., Hamel, F., and Klein, E. K. (2012). Allee effect promotes diversity in traveling waves of colonization. Proceedings of the National Academy of Sciences, 109(23), 8828-8833. doi: https://doi.org/10.1073/pnas.1201695109
| Shifts from pulled to pushed range expansions caused by reduction of landscape connectivity | Maxime Dahirel, Aline Bertin, Marjorie Haond, Aurélie Blin, Eric Lombaert, Vincent Calcagno, Simon Fellous, Ludovic Mailleret, Thibaut Malausa, Elodie Vercken | <p>Range expansions are key processes shaping the distribution of species; their ecological and evolutionary dynamics have become especially relevant today, as human influence reshapes ecosystems worldwide. Many attempts to explain and predict ran... |  | Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology, Experimental Evolution, Phylogeography & Biogeography | Ben Phillips | | 2020-08-04 12:51:56 | View |







