
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields | Recommender▼ | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
15 Sep 2022

Bimodal breeding phenology in the Parsley Frog Pelodytes punctatus as a bet-hedging strategy in an unpredictable environment despite strong priority effectsSpreading the risk of reproductive failure when the environment is unpredictable and ephemeralRecommended by Gabriele Sorci based on reviews by Thomas Haaland and Zoltan RadaiMany species breed in environments that are unpredictable, for instance in terms of the availability of resources needed to raise the offspring. Organisms might respond to such spatial and temporal unpredictability by adopting plastic responses to adjust their reproductive investment according to perceived cues of environmental quality. Some species such as the amphibians might also face the problem of ephemeral habitats, when the ponds where they breed have a chance of drying up before metamorphosis has occurred. In this case, maximizing long-term fitness might involve a strategy of spreading the risk, even though the reproductive success of a single reproductive bout might be lower. Understanding how animals (and plants) get adapted to stochastic environments is particularly crucial in the current context of rapid environmental changes. In this article, Jourdan-Pineau et al. report the results of field surveys of the Parsley Frog (Pelodytes punctatus) in Southern France. This frog has peculiar breeding phenology with females breeding in autumn and spring. The authors provide quite an extensive amount of information on the reproductive success of eggs laid in each season and the possible ecological factors accounting for differences between seasons. Although the presence of interspecific competitors and predators does not seem to account for pond-specific reproductive success, the survival of tadpoles hatching from eggs laid in spring is severely impaired when tadpoles from the autumn cohort have managed to survive. This intraspecific competition takes the form of a “priority” effect where tadpoles from the autumn cohort outcompete the smaller larvae from the spring cohort. Given this strong priority effect, one might tentatively predict that females laying in spring should avoid ponds with tadpoles from the autumn cohort. Surprisingly, however, the authors did not find any evidence for such avoidance, which might indicate strong constraints on the availability of ponds where females might possibly lay. Assuming that each female can indeed lay both in autumn and spring, how is this bimodal phenology maintained? Would not be worthier to allocate all the eggs to the autumn (or the spring) laying season? Eggs laid in autumn and spring have to face different environmental hazards, reducing their hatching success and the probability to produce metamorphs (for instance, tadpoles hatching from eggs laid in autumn have to overwinter which might be a particularly risky phase). Jourdan-Pineau and coworkers addressed this question by adapting a bet-hedging model that was initially developed to investigate the strategy of allocation into seed dormancy of annual plants (Cohen 1966) to the case of the bimodal phenology of the Parsley Frog. By feeding the model with the parameter values obtained from the field surveys, they found that the two breeding strategies (laying in autumn and in spring) can coexist as long as the probability of breeding success in the autumn cohort is between 20% and 80% (the range of values allowing the coexistence of a bimodal phenology shrinking a little bit when considering that frogs can reproduce 5 times during their lifespan instead of three times). This paper provides a very nice illustration of the importance of combining approaches (here field monitoring to gather data that can be used to feed models) to understand the evolution of peculiar breeding strategies. Although future work should attempt to gather individual-based data (in addition to population data), this work shows that spreading the risk can be an adaptive strategy in environments characterized by strong stochastic variation. References Cohen D (1966) Optimizing reproduction in a randomly varying environment. Journal of Theoretical Biology, 12, 119–129. https://doi.org/10.1016/0022-5193(66)90188-3 Jourdan-Pineau H., Crochet P.-A., David P. (2022) Bimodal breeding phenology in the Parsley Frog Pelodytes punctatus as a bet-hedging strategy in an unpredictable environment despite strong priority effects. bioRxiv, 2022.02.24.481784, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.02.24.481784 | Bimodal breeding phenology in the Parsley Frog Pelodytes punctatus as a bet-hedging strategy in an unpredictable environment despite strong priority effects | Helene Jourdan-Pineau, Pierre-Andre Crochet, Patrice David | <p style="text-align: justify;">When environmental conditions are unpredictable, expressing alternative phenotypes spreads the risk of failure, a mixed strategy called bet-hedging. In the southern part of its range, the Parsley Frog <em>Pelodytes ... | | Adaptation, Evolutionary Ecology, Life History | Gabriele Sorci | 2022-02-28 11:53:00 | ||
09 Dec 2019
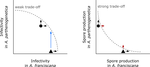
Trait-specific trade-offs prevent niche expansion in two parasitesTrade-offs in fitness components and ecological source-sink dynamics affect host specialisation in two parasites of Artemia shrimpsRecommended by Frédéric Guillaume based on reviews by Anne Duplouy, Seth Barribeau and Cindy Gidoin based on reviews by Anne Duplouy, Seth Barribeau and Cindy Gidoin
Ecological specialisation, especially among parasites infecting a set of host species, is ubiquitous in nature. Host specialisation can be understood as resulting from trade-offs in parasite infectivity, virulence and growth. However, it is not well understood how variation in these trade-offs shapes the overall fitness trade-off a parasite faces when adapting to multiple hosts. For instance, it is not clear whether a strong trade-off in one fitness component may sufficiently constrain the evolution of a generalist parasite despite weak trade-offs in other components. A second mechanism explaining variation in specialisation among species is habitat availability and quality. Rare habitats or habitats that act as ecological sinks will not allow a species to persist and adapt, preventing a generalist phenotype to evolve. Understanding the prevalence of those mechanisms in natural systems is crucial to understand the emergence and maintenance of host specialisation, and biodiversity in general. References [1] Lievens, E.J.P., Michalakis, Y. and Lenormand, T. (2019). Trait-specific trade-offs prevent niche expansion in two parasites. bioRxiv, 621581, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/621581 | Trait-specific trade-offs prevent niche expansion in two parasites | Eva JP Lievens, Yannis Michalakis, Thomas Lenormand | <p>The evolution of host specialization has been studied intensively, yet it is still often difficult to determine why parasites do not evolve broader niches – in particular when the available hosts are closely related and ecologically similar. He... | | Adaptation, Evolutionary Ecology, Evolutionary Epidemiology, Experimental Evolution, Life History, Species interactions | Frédéric Guillaume | 2019-05-13 13:44:34 | ||
31 Mar 2022
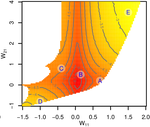
Gene network robustness as a multivariate characterGenetic and environmental robustness are distinct yet correlated evolvable traits in a gene networkRecommended by Frédéric Guillaume based on reviews by Diogo Melo, Charles Mullon and Charles Rocabert
Organisms often show robustness to genetic or environmental perturbations. Whether these two components of robustness can evolve separately is the focus of the paper by Le Rouzic [1]. Using theoretical analysis and individual-based computer simulations of a gene regulatory network model, he shows that multiple aspects of robustness can be investigated as a set of pleiotropically linked quantitative traits. While genetically correlated, various robustness components (e.g., mutational, developmental, homeostasis) of gene expression in the regulatory network evolved more or less independently from each other under directional selection. The quantitative approach of Le Rouzic could explain both how unselected robustness components can respond to selection on other components and why various robustness-related features seem to have their own evolutionary history. Moreover, he shows that all components were evolvable, but not all to the same extent. Robustness to environmental disturbances and gene expression stability showed the largest responses while increased robustness to genetic disturbances was slower. Interestingly, all components were positively correlated and remained so after selection for increased or decreased robustness. This study is an important contribution to the discussion of the evolution of robustness in biological systems. While it has long been recognized that organisms possess the ability to buffer genetic and environmental perturbations to maintain homeostasis (e.g., canalization [2]), the genetic basis and evolutionary routes to robustness and canalization are still not well understood. Models of regulatory gene networks have often been used to address aspects of robustness evolution (e.g., [3]). Le Rouzic [1] used a gene regulatory network model derived from Wagner’s model [4]. The model has as end product the expression level of a set of genes influenced by a set of regulatory elements (e.g., transcription factors). The level and stability of expression are a property of the regulatory interactions in the network. Le Rouzic made an important contribution to the study of such gene regulation models by using a quantitative genetics approach to the evolution of robustness. He crafted a way to assess the mutational variability and selection response of the components of robustness he was interested in. Le Rouzic’s approach opens avenues to investigate further aspects of gene network evolutionary properties, for instance to understand the evolution of phenotypic plasticity. Le Rouzic also discusses ways to measure his different robustness components in empirical studies. As the model is about gene expression levels at a set of protein-coding genes influenced by a set of regulatory elements, it naturally points to the possibility of using RNA sequencing to measure the variation of gene expression in know gene networks and assess their robustness. Robustness could then be studied as a multidimensional quantitative trait in an experimental setting. References [1] Le Rouzic, A (2022) Gene network robustness as a multivariate character. arXiv: 2101.01564, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://arxiv.org/abs/2101.01564 [2] Waddington CH (1942) Canalization of Development and the Inheritance of Acquired Characters. Nature, 150, 563–565. https://doi.org/10.1038/150563a0 [3] Draghi J, Whitlock M (2015) Robustness to noise in gene expression evolves despite epistatic constraints in a model of gene networks. Evolution, 69, 2345–2358. https://doi.org/10.1111/evo.12732 [4] Wagner A (1994) Evolution of gene networks by gene duplications: a mathematical model and its implications on genome organization. Proceedings of the National Academy of Sciences, 91, 4387–4391. https://doi.org/10.1073/pnas.91.10.4387 | Gene network robustness as a multivariate character | Arnaud Le Rouzic | <p style="text-align: justify;">Robustness to genetic or environmental disturbances is often considered as a key property of living systems. Yet, in spite of being discussed since the 1950s, how robustness emerges from the complexity of genetic ar... | | Bioinformatics & Computational Biology, Evolutionary Theory, Genotype-Phenotype, Quantitative Genetics | Frédéric Guillaume | Charles Mullon, Charles Rocabert, Diogo Melo | 2021-01-11 17:48:20 | |
20 Nov 2023
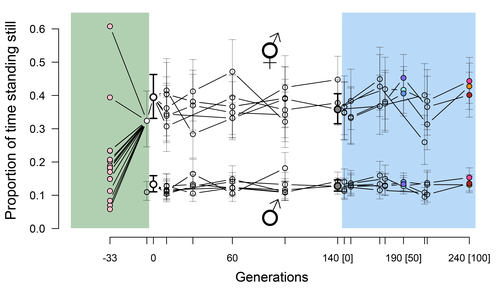
Phenotypic stasis with genetic divergencePhenotypic stasis despite genetic divergence and differentiation in Caenorhabditis elegans.Recommended by Frédéric Guillaume based on reviews by Benoit Pujol and Pedro Simões
Explaining long periods of evolutionary stasis, the absence of change in trait means over geological times, despite the existence of abundant genetic variation in most traits has challenged evolutionary theory since Darwin's theory of evolution by gradual modification (Estes & Arnold 2007). Stasis observed in contemporary populations is even more daunting since ample genetic variation is usually coupled with the detection of selection differentials (Kruuk et al. 2002, Morrissey et al. 2010). Moreover, rapid adaptation to environmental changes in contemporary populations, fuelled by standing genetic variation provides evidence that populations can quickly respond to an adaptive challenge. Explanations for evolutionary stasis usually invoke stabilizing selection as a main actor, whereby optimal trait values remain roughly constant over long periods of time despite small-scale environmental fluctuations. Genetic correlation among traits may also play a significant role in constraining evolutionary changes over long timescales (Schluter 1996). Yet, genetic constraints are rarely so strong as to completely annihilate genetic changes, and they may evolve. Patterns of genetic correlations among traits, as captured in estimates of the G-matrix of additive genetic co-variation, are subject to changes over generations under the action of drift, migration, or selection, among other causes (Arnold et al. 2008). Therefore, under the assumption of stabilizing selection on a set of traits, phenotypic stasis and genetic divergence in patterns of trait correlations may both be observed when selection on trait correlations is weak relative to its effect on trait means. Mallard et al. (2023) set out to test whether selection or drift may explain the divergence in genetic correlation among traits in experimental lines of the nematode Caenorhabditis elegans and whether stabilizing selection may be a driver of phenotypic stasis. To do so, they analyzed the evolution of locomotion behavior traits over 100 generations of lab evolution in a constant and homogeneous environment after 140 generations of domestication from a largely differentiated set of founder populations. The locomotion traits were transition rates between movement states and direction (still, forward or backward movement). They could estimate the traits' broad-sense G-matrix in three populations at two generations (50 and 100), and in the ancestral mixed population. Similarly, they estimated the shape of the selection surface by regressing locomotion behavior on fertility. Armed with both G-matrix and surface estimates, they could test whether the G's orientation matched selection's orientation and whether changes in G were constrained by selection. They found stasis in trait mean over 100 generations but divergence in the amount and orientation of the genetic variation of the traits relative to the ancestral population. The selected populations changed orientation of their G-matrices and lost genetic variation during the experiment in agreement with a model of genetic drift on quantitative traits. Their estimates of selection also point to mostly stabilizing selection on trait combinations with weak evidence of disruptive selection, suggesting a saddle-shaped selection surface. The evolutionary responses of the experimental populations were mostly consistent with small differentiation in the shape of G-matrices during the 100 generations of stabilizing selection. Mallard et al. (2023) conclude that phenotypic stasis was maintained by stabilizing selection and drift in their experiment. They argue that their findings are consistent with a "table-top mountain" model of stabilizing selection, whereby the population is allowed some wiggle room around the trait optimum, leaving space for random fluctuations of trait variation, and especially trait co-variation. The model is an interesting solution that might explain how stasis can be maintained over contemporary times while allowing for random differentiation of trait genetic co-variation. Whether such differentiation can then lead to future evolutionary divergence once replicated populations adapt to a new environment is an interesting idea to follow. References Arnold, S. J., Bürger, R., Hohenlohe, P. A., Ajie, B. C. and Jones, A. G. 2008. Understanding the evolution and stability of the G-matrix. Evolution 62(10): 2451-2461. | Phenotypic stasis with genetic divergence | François Mallard, Luke Noble, Thiago Guzella, Bruno Afonso, Charles F. Baer, Henrique Teotónio | <p style="text-align: justify;">Whether or not genetic divergence in the short-term of tens to hundreds of generations is compatible with phenotypic stasis remains a relatively unexplored problem. We evolved predominantly outcrossing, genetically ... | | Adaptation, Behavior & Social Evolution, Experimental Evolution, Quantitative Genetics | Frédéric Guillaume | 2022-09-01 14:32:53 | ||
12 Apr 2017

POSTPRINT
Genetic drift, purifying selection and vector genotype shape dengue virus intra-host genetic diversity in mosquitoesVectors as motors (of virus evolution)Recommended by Frédéric Fabre and Benoit MouryMany viruses are transmitted by biological vectors, i.e. organisms that transfer the virus from one host to another. Dengue virus (DENV) is one of them. Dengue is a mosquito-borne viral disease that has rapidly spread around the world since the 1940s. One recent estimate indicates 390 million dengue infections per year [1]. As many arthropod-borne vertebrate viruses, DENV has to cross several anatomical barriers in the vector, to multiply in its body and to invade its salivary glands before getting transmissible. As a consequence, vectors are not passive carriers but genuine hosts of the viruses that potentially have important effects on the composition of virus populations and, ultimately, on virus epidemiology and virulence. Within infected vectors, virus populations are expected to acquire new mutations and to undergo genetic drift and selection effects. However, the intensity of these evolutionary forces and the way they shape virus genetic diversity are poorly known. In their study, Lequime et al. [2] finely disentangled the effects of genetic drift and selection on DENV populations during their infectious cycle within mosquito (Aedes aegypti) vectors. They evidenced that the genetic diversity of viruses within their vectors is shaped by genetic drift, selection and vector genotype. The experimental design consisted in artificial acquisition of purified virus by mosquitoes during a blood meal. The authors monitored the diversity of DENV populations in Ae. aegypti individuals at different time points by high-throughput sequencing (HTS). They estimated the intensity of genetic drift and selection effects exerted on virus populations by comparing the DENV diversity at these sampling time points with the diversity in the purified virus stock (inoculum). Disentangling the effects of genetic drift and selection remains a methodological challenge because both evolutionary forces operate concomitantly and both reduce genetic diversity. However, selection reduces diversity in a reproducible manner among experimental replicates (here, mosquito individuals): the fittest variants are favoured at the expense of the weakest ones. In contrast, genetic drift reduces diversity in a stochastic manner among replicates. Genetic drift acts equally on all variants irrespectively of their fitness. The strength of genetic drift is frequently evaluated with the effective population size Ne: the lower Ne, the stronger the genetic drift [3]. The estimation of the effective population size of DENV populations by Lequime et al. [2] was based on single-nucleotide polymorphisms (SNPs) that were (i) present both in the inoculum and in the virus populations sampled at the different time points and (ii) that were neutral (or nearly-neutral) and therefore subjected to genetic drift only and insensitive to selection. As expected for viruses that possess small and constrained genomes, such neutral SNPs are extremely rare. Starting from a set of >1800 SNPs across the DENV genome, only three SNPs complied with the neutrality criteria and were enough represented in the sequence dataset for a precise Ne estimation. Using the method described by Monsion et al. [4], Lequime et al. [2] estimated Ne values ranging from 5 to 42 viral genomes (95% confidence intervals ranged from 2 to 161 founding viral genomes). Consequently, narrow bottlenecks occurred at the virus acquisition step, since the blood meal had allowed the ingestion of ca. 3000 infectious virus particles, on average. Interestingly, bottleneck sizes did not differ between mosquito genotypes. Monsion et al.’s [4] formula provides only an approximation of Ne. A corrected formula has been recently published [5]. We applied this exact Ne formula to the means and variances of the frequencies of the three neutral markers estimated before and after the bottlenecks (Table 1 in [2]), and nearly identical Ne estimates were obtained with both formulas. Selection intensity was estimated from the dN/dS ratio between the nonsynonymous and synonymous substitution rates using the HTS data on DENV populations. DENV genetic diversity increased following initial infection but was restricted by strong purifying selection during virus expansion in the midgut. Again, no differences were detected between mosquito genotypes. However and importantly, significant differences in DENV genetic diversity were detected among mosquito genotypes. As they could not be related to differences in initial genetic drift or to selection intensity, the authors raise interesting alternative hypotheses, including varying rates of de novo mutations due to differences in replicase fidelity or differences in the balancing selection regime. Interestingly, they also suggest that this observation could simply result from a methodological issue linked to the detection threshold of low-frequency SNPs. References [1] Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, et al. 2013. The global distribution and burden of dengue. Nature 496: 504–7 doi: 10.1038/nature12060 [2] Lequime S, Fontaine A, Gouilh MA, Moltini-Conclois I and Lambrechts L. 2016. Genetic drift, purifying selection and vector genotype shape dengue virus intra-host genetic diversity in mosquitoes. PloS Genetics 12: e1006111 doi: 10.1371/journal.pgen.1006111 [3] Charlesworth B. 2009. Effective population size and patterns of molecular evolution and variation. Nature Reviews Genetics 10: 195-205 doi: 10.1038/nrg2526 [4] Monsion B, Froissart R, Michalakis Y and Blanc S. 2008. Large bottleneck size in cauliflower mosaic virus populations during host plant colonization. PloS Pathogens 4: e1000174 doi: 10.1371/journal.ppat.1000174 [5] Thébaud G and Michalakis Y. 2016. Comment on ‘Large bottleneck size in cauliflower mosaic virus populations during host plant colonization’ by Monsion et al. (2008). PloS Pathogens 12: e1005512 doi: 10.1371/journal.ppat.1005512 | Genetic drift, purifying selection and vector genotype shape dengue virus intra-host genetic diversity in mosquitoes | Lequime S, Fontaine A, Gouilh MA, Moltini-Conclois I and Lambrechts L | Due to their error-prone replication, RNA viruses typically exist as a diverse population of closely related genomes, which is considered critical for their fitness and adaptive potential. Intra-host demographic fluctuations that stochastically re... | | Evolutionary Dynamics, Molecular Evolution, Population Genetics / Genomics | Frédéric Fabre | 2017-04-10 14:26:04 | ||
29 Sep 2022
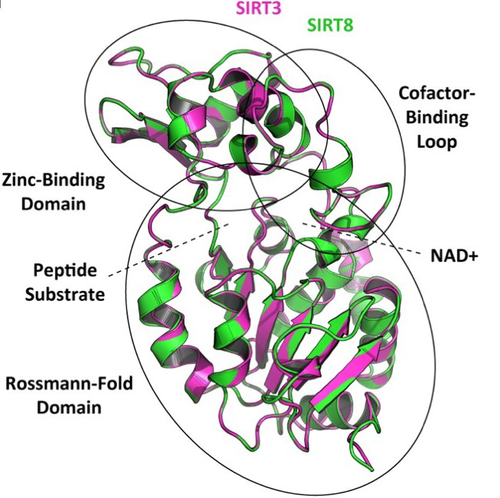
How many sirtuin genes are out there? evolution of sirtuin genes in vertebrates with a description of a new family memberMaking sense of vertebrate sirtuin genesRecommended by Frédéric Delsuc based on reviews by Filipe Castro, Nicolas Leurs and 1 anonymous reviewerSirtuin proteins are class III histone deacetylases that are involved in a variety of fundamental biological functions mostly related to aging. These proteins are located in different subcellular compartments and are associated with different biological functions such as metabolic regulation, stress response, and cell cycle control [1]. In mammals, the sirtuin gene family is composed of seven paralogs (SIRT1-7) grouped into four classes [2]. Due to their involvement in maintaining cell cycle integrity, sirtuins have been studied as a way to understand fundamental mechanisms governing longevity [1]. Indeed, the downregulation of sirtuin genes with aging seems to explain much of the pathophysiology that accumulates with aging [3]. Biomedical studies have thus explored the potential therapeutic implications of sirtuins [4] but whether they can effectively be used as molecular targets for the treatment of human diseases remains to be demonstrated [1]. Despite this biomedical interest and some phylogenetic analyses of sirtuin paralogs mostly conducted in mammals, a comprehensive evolutionary analysis of the sirtuin gene family at the scale of vertebrates was still lacking. In this preprint, Opazo and collaborators [5] took advantage of the increasing availability of whole-genome sequences for species representing all main groups of vertebrates to unravel the evolution of the sirtuin gene family. To do so, they undertook a phylogenomic approach in its original sense aimed at improving functional predictions by evolutionary analysis [6] in order to inventory the full vertebrate sirtuin gene repertoire and reconstruct its precise duplication history. Harvesting genomic databases, they extracted all predicted sirtuin proteins and performed phylogenetic analyses based on probabilistic inference methods. Maximum likelihood and Bayesian analyses resulted in well-resolved and congruent phylogenetic trees dividing vertebrate sirtuin genes into three major clades. These analyses also revealed an additional eighth paralog that was previously overlooked because of its restricted phyletic distribution. This newly identified sirtuin family member (named SIRT8) was recovered with unambiguous statistical support as a sister-group to the SIRT3 clade. Comparative genomic analyses based on conserved gene synteny confirmed that SIRT8 was present in all sampled non-amniote vertebrate genomes (cartilaginous fish, bony fish, coelacanth, lungfish, and amphibians) except cyclostomes. SIRT8 has thus most likely been lost in the last common ancestor of amniotes (mammals, reptiles, and birds). Discovery of such previously unknown genes in vertebrates is not completely surprising given the plethora of high-quality genomes now available. However, this study highlights the importance of considering a broad taxonomic sampling to infer evolutionary patterns of gene families that have been mostly studied in mammals because of their potential importance for human biology. Based on its phylogenetic position as closely related to SIRT3 within class I, it could be predicted that the newly identified SIRT8 paralog likely has a deacetylase activity and is probably located in mitochondria. To test these evolutionary predictions, Opazo and collaborators [5] conducted further bioinformatics analyses and functional experiments using the elephant shark (Callorhinchus milii) as a model species. RNAseq expression data were analyzed to determine tissue-specific transcription of sirtuin genes in vertebrates, including SIRT8 found to be mainly expressed in the ovary, which suggests a potential role in biological processes associated with reproduction. The elephant shark SIRT8 protein sequence was used with other vertebrates for comparative analyses of protein structure modeling and subcellular localization prediction both pointing to a probable mitochondrial localization. The protein localization and its function were further characterized by immunolocalization in transfected cells, and enzymatic and functional assays, which all confirmed the prediction that SIRT8 proteins are targeted to the mitochondria and have deacetylase activity. The extensive experimental efforts made in this study to shed light on the function of this newly discovered gene are both rare and highly commendable. Overall, this work by Opazo and collaborators [5] provides a comprehensive phylogenomic study of the sirtuin gene family in vertebrates based on detailed evolutionary analyses using state-of-the-art phylogenetic reconstruction methods. It also illustrates the power of adopting an integrative comparative approach supplementing the reconstruction of the duplication history of the gene family with complementary functional experiments in order to elucidate the function of the newly discovered SIRT8 family member. These results provide a reference phylogenetic framework for the evolution of sirtuin genes and the further functional characterization of the eight vertebrate paralogs with potential relevance for understanding the cellular biology of aging and its associated diseases in human. References [1] Vassilopoulos A, Fritz KS, Petersen DR, Gius D (2011) The human sirtuin family: Evolutionary divergences and functions. Human Genomics, 5, 485. https://doi.org/10.1186/1479-7364-5-5-485 [2] Yamamoto H, Schoonjans K, Auwerx J (2007) Sirtuin Functions in Health and Disease. Molecular Endocrinology, 21, 1745–1755. https://doi.org/10.1210/me.2007-0079 [3] Morris BJ (2013) Seven sirtuins for seven deadly diseases ofaging. Free Radical Biology and Medicine, 56, 133–171. https://doi.org/10.1016/j.freeradbiomed.2012.10.525 [4] Bordo D Structure and Evolution of Human Sirtuins. Current Drug Targets, 14, 662–665. http://dx.doi.org/10.2174/1389450111314060007 [5] Opazo JC, Vandewege MW, Hoffmann FG, Zavala K, Meléndez C, Luchsinger C, Cavieres VA, Vargas-Chacoff L, Morera FJ, Burgos PV, Tapia-Rojas C, Mardones GA (2022) How many sirtuin genes are out there? evolution of sirtuin genes in vertebrates with a description of a new family member. bioRxiv, 2020.07.17.209510, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.07.17.209510 [6] Eisen JA (1998) Phylogenomics: Improving Functional Predictions for Uncharacterized Genes by Evolutionary Analysis. Genome Research, 8, 163–167. https://doi.org/10.1101/gr.8.3.163 | How many sirtuin genes are out there? evolution of sirtuin genes in vertebrates with a description of a new family member | Juan C. Opazo, Michael W. Vandewege, Federico G. Hoffmann, Kattina Zavala, Catalina Meléndez, Charlotte Luchsinger, Viviana A. Cavieres, Luis Vargas-Chacoff, Francisco J. Morera, Patricia V. Burgos, Cheril Tapia-Rojas, Gonzalo A. Mardones | <p style="text-align: justify;">Studying the evolutionary history of gene families is a challenging and exciting task with a wide range of implications. In addition to exploring fundamental questions about the origin and evolution of genes, disent... | | Molecular Evolution | Frédéric Delsuc | Filipe Castro, Anonymous, Nicolas Leurs | 2022-05-12 16:06:04 | |
04 Mar 2021
Simulation of bacterial populations with SLiMSimulating bacterial evolution forward-in-timeRecommended by Frederic Bertels based on reviews by 3 anonymous reviewersJean Cury and colleagues (2021) have developed a protocol to simulate bacterial evolution in SLiM. In contrast to existing methods that depend on the coalescent, SLiM simulates evolution forward in time. SLiM has, up to now, mostly been used to simulate the evolution of eukaryotes (Haller and Messer 2019), but has been adapted here to simulate evolution in bacteria. Forward-in-time simulations are usually computationally very costly. To circumvent this issue, bacterial population sizes are scaled down. One would now expect results to become inaccurate, however, Cury et al. show that scaled-down forwards simulations provide very accurate results (similar to those provided by coalescent simulators) that are consistent with theoretical expectations. Simulations were analyzed and compared to existing methods in simple and slightly more complex scenarios where recombination affects evolution. In all scenarios, simulation results from coalescent methods (fastSimBac (De Maio and Wilson 2017), ms (Hudson 2002)) and scaled-down forwards simulations were very similar, which is very good news indeed. A biologist not aware of the complexities of forwards, backwards simulations and the coalescent, might now naïvely ask why another simulation method is needed if existing methods perform just as well. To address this question the manuscript closes with a very neat example of what exactly is possible with forwards simulations that cannot be achieved using existing methods. The situation modeled is the growth and evolution of a set of 50 bacteria that are randomly distributed on a petri dish. One side of the petri dish is covered in an antibiotic the other is antibiotic-free. Over time, the bacteria grow and acquire antibiotic resistance mutations until the entire artificial petri dish is covered with a bacterial lawn. This simulation demonstrates that it is possible to simulate extremely complex (e.g. real world) scenarios to, for example, assess whether certain phenomena are expected with our current understanding of bacterial evolution, or whether there are additional forces that need to be taken into account. Hence, forwards simulators could significantly help us to understand what current models can and cannot explain in evolutionary biology.
References Cury J, Haller BC, Achaz G, Jay F (2021) Simulation of bacterial populations with SLiM. bioRxiv, 2020.09.28.316869, version 5 peer-reviewed and recommended by Peer community in Evolutionary Biology. https://doi.org/10.1101/2020.09.28.316869 De Maio N, Wilson DJ (2017) The Bacterial Sequential Markov Coalescent. Genetics, 206, 333–343. https://doi.org/10.1534/genetics.116.198796 Haller BC, Messer PW (2019) SLiM 3: Forward Genetic Simulations Beyond the Wright–Fisher Model. Molecular Biology and Evolution, 36, 632–637. https://doi.org/10.1093/molbev/msy228 Hudson RR (2002) Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics, 18, 337–338. https://doi.org/10.1093/bioinformatics/18.2.337 | Simulation of bacterial populations with SLiM | Jean Cury, Benjamin C. Haller, Guillaume Achaz, and Flora Jay | <p>Simulation of genomic data is a key tool in population genetics, yet, to date, there is no forward-in-time simulator of bacterial populations that is both computationally efficient and adaptable to a wide range of scenarios. Here we demonstrate... | | Bioinformatics & Computational Biology, Population Genetics / Genomics | Frederic Bertels | 2020-10-02 19:03:42 | ||
23 Jan 2020

A novel workflow to improve multi-locus genotyping of wildlife species: an experimental set-up with a known model systemImproving the reliability of genotyping of multigene families in non-model organismsRecommended by François Rousset based on reviews by Sebastian Ernesto Ramos-Onsins, Helena Westerdahl and Thomas BigotThe reliability of published scientific papers has been the topic of much recent discussion, notably in the biomedical sciences [1]. Although small sample size is regularly pointed as one of the culprits, big data can also be a concern. The advent of high-throughput sequencing, and the processing of sequence data by opaque bioinformatics workflows, mean that sequences with often high error rates are produced, and that exact but slow analyses are not feasible. References [1] Ioannidis, J. P. A, Greenland, S., Hlatky, M. A., Khoury, M. J., Macleod, M. R., Moher, D., Schulz, K. F. and Tibshirani, R. (2014) Increasing value and reducing waste in research design, conduct, and analysis. The Lancet, 383, 166-175. doi: 10.1016/S0140-6736(13)62227-8 | A novel workflow to improve multi-locus genotyping of wildlife species: an experimental set-up with a known model system | Gillingham, Mark A. F., Montero, B. Karina, Wilhelm, Kerstin, Grudzus, Kara, Sommer, Simone and Santos, Pablo S. C. | <p>Genotyping novel complex multigene systems is particularly challenging in non-model organisms. Target primers frequently amplify simultaneously multiple loci leading to high PCR and sequencing artefacts such as chimeras and allele amplification... | | Bioinformatics & Computational Biology, Evolutionary Ecology, Genome Evolution, Molecular Evolution | François Rousset | Helena Westerdahl, Sebastian Ernesto Ramos-Onsins, Paul J. McMurdie , Arnaud Estoup, Vincent Segura, Jacek Radwan , Torbjørn Rognes , William Stutz , Kevin Vanneste , Thomas Bigot, Jill A. Hollenbach , Wieslaw Babik , Marie-Christin... | 2019-05-15 17:30:44 | |
03 Oct 2023
The evolutionary dynamics of plastic foraging and its ecological consequences: a resource-consumer modelEvolution and consequences of plastic foraging behavior in consumer-resource ecosystemsRecommended by François Rousset based on reviews by 2 anonymous reviewersPlastic responses of organisms to their environment may be maladaptive in particular when organisms are exposed to new environments. Phenotypic plasticity may also have opposite effects on the adaptive response of organisms to environmental changes: whether phenotypic plasticity favors or hinders such adaptation depends on a balance between the ability of the population to respond to the change non-genetically in the short term, and the weakened genetic response to environmental change. These topics have received continued attention, particularly in the context of climate change (e.g., Chevin et al. 2013, Duputié et al., 2015, Vinton et al . 2022). In their work, Ledru et al. focus on the adaptive nature of plastic behavior and on its consequences in a consumer-resource ecosystem. As they emphasize, previous works have found that plastic foraging promotes community stability, but these did not consider plasticity as an evolving trait, so Ledru et al. set out to test whether this conclusion holds when both plastic foraging and niche traits of consumers and resources evolve (though ultimately, their new conclusions may not all depend on plasticity evolving). Along the way, they first seek to clarify when such plasticity will evolve, and how it affects the evolution of the niche diversity of consumers and resources, before turning to the question of consumer persistence. The model is rather complex, as three traits are allowed to evolve, and the resource uptake expressed through plastic behavior has its own dynamics affected by some form of social learning. Classically, in models of niche evolution, a consumer's efficiency in exploiting a resource characterized by a trait y (here, the resource's individual niche trait), has been described in terms of location-scale (typically Gaussian) kernels, with mean x (the consumer's individual niche trait) specifying the most efficiently exploited resource, and with variance characterizing individual niche breadth. The evolution of the variance has been considered in some previous models but is assumed to be fixed here. Rather, the new model considers the evolution of the distribution of resource traits, of the consumer's individual niche trait (which is not plastic), and of a "plastic foraging trait" that controls the relative time spent foraging plastically versus foraging randomly. When foraging plastically, the consumers modify their foraging effort towards the type of resource that maximizes their energy intake. in some previous models, the effect of variation in the extent of plastic foraging was already considered, but the evolution of allocation to a plastic foraging strategy versus random foraging was not considered. The model is formulated through reaction-diffusion equations, and its dynamics is investigated by numerical integration. Foraging plasticity readily evolves, when resources vary widely enough, competition for resources is strong, and the cost of plasticity is weak. This means in particular that a large individual niche width of consumers selects for increased plastic foraging, as the evolution of plastic foraging leads to reduced niche overlap between consumers. The evolution of plastic foraging itself generally, though not always, favors the diversification of the niche traits of consumers and of resources. There is thus a positive feedback loop between plastic foraging and resource diversity. Ledru et al. conclude that the total niche width of the consumer population should also correlate with the evolution of plastic foraging, an implication which they relate to the so-called niche variation hypothesis and to empirical tests of it. The joint evolution of the consumer's individual niche trait and plastic foraging trait generates a striking pattern within populations: consumers whose individual niche trait is at an edge of the resource distribution forage more plastically. The authors observe that this relatively simple prediction has not been subjected to any empirical test. Returning to the question of consumer persistence, Ledru et al. evaluate this persistence when consumer mortality increases, and in response to either gradual or sudden environmental changes. These different perturbations all reduce the benefits of plastic foraging. The effect of plastic foraging on stability are complex, being negative or positive effect depending on the type of disturbance, and in particular the ecosystem has a lower sustainable rate of environmental change in the presence of plastic foraging. However, allowing the evolutionary regression of plastic foraging then has a comparatively positive effect on persistence. Despite the substantial effort devoted to analyzing this complex model, relaxing some of its assumptions would likely reveal further complexities. Notably, the overall effect of plasticity on consumer persistence depends on effects already encountered in models of the adaptive response of single species to environmental change: a fast non-genetic response in the short term versus a weakened genetic response in the longer term. The overall balance between these opposite effects on adaptation may be difficult to predict robustly. In the case of a constant rate of environmental change, the results of the present model depend on a lag load between the trait changes of consumer and resource populations, and the extent of the lag may also depend on many factors, such as the extent of genetic variation (e.g., Bürger & Lynch, 1995) for niche traits in consumers and resources. Here, the same variance of mutational effects was assumed for all three evolving traits. Further, spatial environmental variation, a central issue in studies of adaptive responses to environmental changes (e.g., Parmesan, 2006, Zhu et al., 2012), was not considered. Finally, the rate of adjustment of effort by consumers with given niche trait and plastic foraging trait values was assumed proportional to the density of consumers with such trait values. This was justified as a way of accounting for the use of social cues during foraging, but to the extent that they occur, social effects could manifest themselves through other learning dynamics. In conclusion, Ledru et al. have addressed a broad range of questions, suggesting new empirical tests of behavioural patterns on one side, and recovering in the context of community response to environmental changes a complexity that could be expected from earlier works on adaptive responses of organisms but that has been overlooked by previous works on community effects of phenotypic plasticity. References Bürger, R. and Lynch, M. (1995), Evolution and extinction in a changing environment: a quantitative-genetic analysis. Evolution, 49: 151-163. https://doi.org/10.1111/j.1558-5646.1995.tb05967.x Chevin, L.-M., Collins, S. and Lefèvre, F. (2013), Phenotypic plasticity and evolutionary demographic responses to climate change: taking theory out to the field. Funct Ecol, 27: 967-979. https://doi.org/10.1111/j.1365-2435.2012.02043.x Duputié, A., Rutschmann, A., Ronce, O. and Chuine, I. (2015), Phenological plasticity will not help all species adapt to climate change. Glob Change Biol, 21: 3062-3073. https://doi-org.inee.bib.cnrs.fr/10.1111/gcb.12914 Ledru, L., Garnier, J., Guillot, O., Faou, E., & Ibanez, S. (2023). The evolutionary dynamics of plastic foraging and its ecological consequences: a resource-consumer model. EcoEvoRxiv, ver. 4 peer-reviewed and recommended by Peer Community In Evolutionary Biology. https://doi.org/10.32942/X2QG7M Parmesan, C. (2006) Ecological and evolutionary responses to recent climate change Vinton, A.C., Gascoigne, S.J.L., Sepil, I., Salguero-Gómez, R., (2022) Plasticity’s role in adaptive evolution depends on environmental change components. Trends in Ecology & Evolution, 37: 1067-1078. Zhu, K., Woodall, C.W. and Clark, J.S. (2012), Failure to migrate: lack of tree range expansion in response to climate change. Glob Change Biol, 18: 1042-1052. https://doi.org/10.1111/j.1365-2486.2011.02571.x | The evolutionary dynamics of plastic foraging and its ecological consequences: a resource-consumer model | Léo Ledru, Jimmy Garnier, Océane Guillot, Erwan Faou, Camille Noûs, Sébastien Ibanez | <p style="text-align: justify;">Phenotypic plasticity has important ecological and evolutionary consequences. In particular, behavioural phenotypic plasticity such as plastic foraging (PF) by consumers, may enhance community stability. Yet little ... | | Bioinformatics & Computational Biology, Evolutionary Dynamics, Evolutionary Ecology, Phenotypic Plasticity | François Rousset | 2023-03-25 12:04:08 | ||
11 Jul 2022

Mutualists construct the ecological conditions that trigger the transition from parasitismGive them some space: how spatial structure affects the evolutionary transition towards mutualistic symbiosisRecommended by Francois Massol based on reviews by Eva Kisdi and 3 anonymous reviewers
The evolution of mutualistic symbiosis is a puzzle that has fascinated evolutionary ecologist for quite a while. Data on transitions between symbiotic bacterial ways of life has evidenced shifts from mutualism towards parasitism and vice versa (Sachs et al., 2011), so there does not seem to be a strong determinism on those transitions. From the host’s perspective, mutualistic symbiosis implies at the very least some form of immune tolerance, which can be costly (e.g. Sorci, 2013). Empirical approaches thus raise very important questions: How can symbiosis turn from parasitism into mutualism when it seemingly needs such a strong alignment of selective pressures on both the host and the symbiont? And yet why is mutualistic symbiosis so widespread and so important to the evolution of macro-organisms (Margulis, 1998)? While much of the theoretical literature on the evolution of symbiosis and mutualism has focused on either the stability of such relationships when non-mutualists can invade the host-symbiont system (e.g. Ferrière et al., 2007) or the effect of the mode of symbiont transmission on the evolutionary dynamics of mutualism (e.g. Genkai-Kato and Yamamura, 1999), the question remains whether and under which conditions parasitic symbiosis can turn into mutualism in the first place. Earlier results suggested that spatial demographic heterogeneity between host populations could be the leading determinant of evolution towards mutualism or parasitism (Hochberg et al., 2000). Here, Ledru et al. (2022) investigate this question in an innovative way by simulating host-symbiont evolutionary dynamics in a spatially explicit context. Their hypothesis is intuitive but its plausibility is difficult to gauge without a model: Does the evolution towards mutualism depend on the ability of the host and symbiont to evolve towards close-range dispersal in order to maintain clusters of efficient host-symbiont associations, thus outcompeting non-mutualists? I strongly recommend reading this paper as the results obtained by the authors are very clear: competition strength and the cost of dispersal both affect the likelihood of the transition from parasitism to mutualism, and once mutualism has set in, symbiont trait values clearly segregate between highly dispersive parasites and philopatric mutualists. The demonstration of the plausibility of their hypothesis is accomplished with brio and thoroughness as the authors also examine the conditions under which the transition can be reversed, the impact of the spatial range of competition and the effect of mortality. Since high dispersal cost and strong, long-range competition appear to be the main factors driving the evolutionary transition towards mutualistic symbiosis, now is the time for empiricists to start investigating this question with spatial structure in mind. References Ferrière, R., Gauduchon, M. and Bronstein, J. L. (2007) Evolution and persistence of obligate mutualists and exploiters: competition for partners and evolutionary immunization. Ecology Letters, 10, 115-126. https://doi.org/10.1111/j.1461-0248.2006.01008.x Genkai-Kato, M. and Yamamura, N. (1999) Evolution of mutualistic symbiosis without vertical transmission. Theoretical Population Biology, 55, 309-323. https://doi.org/10.1006/tpbi.1998.1407 Hochberg, M. E., Gomulkiewicz, R., Holt, R. D. and Thompson, J. N. (2000) Weak sinks could cradle mutualistic symbioses - strong sources should harbour parasitic symbioses. Journal of Evolutionary Biology, 13, 213-222. https://doi.org/10.1046/j.1420-9101.2000.00157.x Ledru L, Garnier J, Rohr M, Noûs C and Ibanez S (2022) Mutualists construct the ecological conditions that trigger the transition from parasitism. bioRxiv, 2021.08.18.456759, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.08.18.456759 Margulis, L. (1998) Symbiotic planet: a new look at evolution, Basic Books, Amherst. Sachs, J. L., Skophammer, R. G. and Regus, J. U. (2011) Evolutionary transitions in bacterial symbiosis. Proceedings of the National Academy of Sciences, 108, 10800-10807. https://doi.org/10.1073/pnas.1100304108 Sorci, G. (2013) Immunity, resistance and tolerance in bird–parasite interactions. Parasite Immunology, 35, 350-361. https://doi.org/10.1111/pim.12047 | Mutualists construct the ecological conditions that trigger the transition from parasitism | Leo Ledru, Jimmy Garnier, Matthias Rohr, Camille Nous, Sebastien Ibanez | <p>The evolution of mutualism between hosts and initially parasitic symbionts represents a major transition in evolution. Although vertical transmission of symbionts during host reproduction and partner control both favour the stability of mutuali... | | Evolutionary Ecology, Species interactions | Francois Massol | 2021-08-20 12:25:40 |




