
Latest recommendations

| Id | Title | Authors | Abstract | Picture | Thematic fields | Recommender▼ | Reviewers | Submission date | |
|---|---|---|---|---|---|---|---|---|---|
16 Mar 2017

POSTPRINT
Correlated paternity measures mate monopolization and scales with the magnitude of sexual selectionMeasurement of sexual selection in plants made easierRecommended by Emmanuelle Porcher and Mathilde DufaySexual selection occurs in flowering plants too. However it tends to be understudied in comparison to animal sexual selection, in part because the minuscule size and long dispersal distances of the individuals producing male gametes (pollen grains) seriously complicate the estimation of male siring success and thereby the measurement of sexual selection. Dorken and Perry [1] introduce a novel and clever approach to estimate sexual selection in plants, which bypasses the need for a direct quantification of absolute male mating success. This approach builds on the fact that the strength of sexual selection is directly related to the ability of individuals to monopolize mates [2]. In plants, mate monopolization can be assessed by examining the proportion of seeds produced by a given plant that are full-sibs, i.e. that share the same father. A nice feature of this proportion of full-sib seeds per maternal parent is it equals the coefficient of correlated paternity of Ritland [3], which can be readily obtained from the hundreds of plant mating system studies using genetic markers. A less desirable feature of the proportion of full sibs per maternal plant is that it is inversely related to population size, an effect that should be corrected for. The resulting index of mate monopolization is a simple product: (coefficient of correlated paternity)x(population size – 1). The authors test whether their index of mate monopolization is a good correlate of sexual selection, measured more traditionally as the selection differential on a trait influencing mating success, using a combination of theoretical and experimental approaches. Both approaches confirm that the two quantities are positively correlated, which suggests that the index of mate monopolization could be a convenient way to estimate the relative strength of sexual selection in flowering plants. These results call for further investigation, e.g. to verify that the effect of population size is well controlled for, or to assess the effects of non-random mating and inbreeding depression; however, this work paves the way for an expansion of sexual selection studies in flowering plants. References [1] Dorken ME and Perry LE. 2017. Correlated paternity measures mate monopolization and scales with the magnitude of sexual selection. Journal of Evolutionary Biology 30: 377-387 doi: 10.1111/jeb.13013 [2] Klug H, Heuschele J, Jennions M and Kokko H. 2010. The mismeasurement of sexual selection. Journal of Evolutionary Biology 23:447-462. doi: 10.1111/j.1420-9101.2009.01921.x [3] Ritland K. 1989. Correlated matings in the partial selfer Mimulus guttatus. Evolution 43:848-859. doi: 10.2307/2409312 | Correlated paternity measures mate monopolization and scales with the magnitude of sexual selection | Dorken, ME and Perry LE | Indirect measures of sexual selection have been criticized because they can overestimate the magnitude of selection. In particular, they do not account for the degree to which mating opportunities can be monopolized by individuals of the sex that ... | | Sexual Selection | Emmanuelle Porcher | 2017-03-13 23:22:26 | ||
14 May 2020

Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisonsFrom populations to subspecies… to species? Contrasting patterns of local adaptation in closely-related taxa and their potential contribution to species divergenceRecommended by Emmanuelle Porcher based on reviews by Sophie Karrenberg, Santiago C. Gonzalez-Martinez and 1 anonymous reviewerElevation gradients are convenient and widely used natural setups to study local adaptation, particularly in these times of rapid climate change [e.g. 1]. Marin and her collaborators [2] did not follow the mainstream, however. Instead of tackling adaptation to climate change, they used elevation gradients to address another crucial evolutionary question [3]: could adaptation to altitude lead to ecological speciation, i.e. reproductive isolation between populations in spite of gene flow? More specifically, they examined how much local adaptation to environmental variation differed among closely-related, recently diverged subspecies. They studied several populations of two subspecies of snapdragon (Antirrhinum majus), with adjacent geographical distributions. Using common garden experiments and the classical, but still useful, QST-FST comparison, they demonstrate contrasting patterns of local adaptation to altitude between the two subspecies, with several traits under divergent selection in A. majus striatum but none in A. majus pseudomajus. These differences in local adaptation may contribute to species divergence, and open many stimulating questions on the underlying mechanisms, such as the identity of environmental drivers or contribution of reproductive isolation involving flower color polymorphism. References [1] Anderson, J. T., and Wadgymar, S. M. (2020). Climate change disrupts local adaptation and favours upslope migration. Ecology letters, 23(1), 181-192. doi: 10.1111/ele.13427 | Potential adaptive divergence between subspecies and populations of snapdragon plants inferred from QST – FST comparisons | Sara Marin, Anaïs Gibert, Juliette Archambeau, Vincent Bonhomme, Mylène Lascoste and Benoit Pujol | <p>Phenotypic divergence among natural populations can be explained by natural selection or by neutral processes such as drift. Many examples in the literature compare putatively neutral (FST) and quantitative genetic (QST) differentiation in mult... | | Adaptation, Evolutionary Ecology, Genotype-Phenotype, Morphological Evolution, Quantitative Genetics | Emmanuelle Porcher | 2018-08-05 15:34:30 | ||
13 Dec 2016

POSTPRINT
Repeated replacements of an intrabacterial symbiont in the tripartite nested mealybug symbiosisObligate dependence does not preclude changing partners in a Russian dolls symbiotic systemRecommended by Emmanuelle Jousselin and Fabrice VavreSymbiotic associations with bacterial partners have facilitated important evolutionary transitions in the life histories of eukaryotes. For instance, many insects have established long-term interactions with intracellular bacteria that provide them with essential nutrients lacking in their diet. However, despite the high level of interdependency among organisms involved in endosymbiotic systems, examples of symbiont replacements along the evolutionary history of insect hosts are numerous.
In their paper, Husnik and McCutcheon [1] test the stability of symbiotic systems in a particularly imbricated Russian-doll type interaction, where one bacterium lives insides another bacterium, which itself lives inside insect cells. For their study, they chose representative species of mealybugs (Pseudococcidae), a species rich group of sap-feeding insects that hosts diverse and complex symbiotic systems. In species of the subfamily Pseudococcinae, data published so far suggest that the primary symbiont, a ß-proteobacterium named Tremblaya princeps, is supplemented by a second bacterial symbiont (a ϒ-proteobaterium) that lives within its cytoplasm; both participate to the metabolic pathways that provide essential amino acids and vitamins to their hosts. Here, Husnik and McCutcheon generate host and endosymbiont genome data for five phylogenetically divergent species of Pseudococcinae in order to better understand: 1) the evolutionary history of the symbiotic associations; 2) the metabolic roles of each partner, 3) the timing and origin of Horizontal Gene Transfers (HGT) between the hosts and their symbionts. Reference [1] Husnik F., McCutcheon JP. 2016. Repeated replacements of an intrabacterial symbiont in the tripartite nested mealybug symbiosis. PNAS 113: E5416-E5424. doi: 10.1073/pnas.1603910113 | Repeated replacements of an intrabacterial symbiont in the tripartite nested mealybug symbiosis | Husnik F, McCutcheon JP | Stable endosymbiosis of a bacterium into a host cell promotes cellular and genomic complexity. The mealybug *Planococcus citri* has two bacterial endosymbionts with an unusual nested arrangement: the γ-proteobacterium *Moranella endobia* lives in ... | | Phylogenetics / Phylogenomics, Species interactions | Emmanuelle Jousselin | 2016-12-13 14:27:09 | ||
02 May 2023
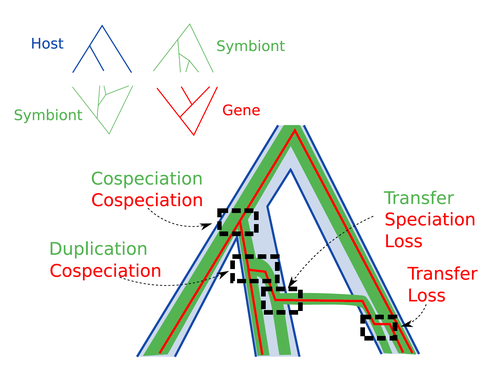
Host-symbiont-gene phylogenetic reconciliationReconciling molecular evolution and evolutionary ecology studies: a phylogenetic reconciliation method for gene-symbiont-host systemsRecommended by Emmanuelle Jousselin based on reviews by Vincent Berry and Catherine MatiasInteractions between species are a driving force in evolution. Many organisms host symbiotic partners that live all or part of their life in or on their host. Whether they are mutualistic or parasitic, these symbiotic associations impose strong selective pressures on both partners and affect their evolutionary trajectories. In fine, they can have a significant impact on the diversification patterns of both host and symbiont lineages, with symbiotic lineages sometimes speciating simultaneously with their hosts and/or switching from one host species to another. Long-term associations between species can also result in gene transfers between the involved organisms. Those lateral gene transfers are a source of ecological innovation but can obscure the phylogenetic signals and render the process of phylogenetic reconstructions complex (Lerat et al. 2003). Methods known as reconciliations explore similarities and differences between phylogenetic trees. They have been widely used to both compare the diversification patterns of hosts and symbionts and identify lateral gene transfers between species. Though the reconciliation approaches used in host/ symbiont and species/ gene phylogenetic studies are identical, they are always applied separately to solve either molecular evolution questions or investigate the evolution of ecological interactions. However, the two questions are often intimately linked and the current interest in multi-level systems (e.g. the holobiont concept) calls for a unique model that will take into account three-level nested organization (gene/symbiont/ host) where both symbiont and genes can transfer among hosts. Here Menet and collaborators (2023) provide such a model to produce three-level reconciliations. In order to do so, they extend the two-level reconciliation model implemented in “ALE” software (Szöllősi et al. 2013), one of the most used and proven reconciliation methods. Briefly, given a symbiont gene tree, a symbiont tree and a host tree, as in previous reconciliation models, the symbiont tree is mapped onto the host tree by mixing three types of events: Duplication, Transfer or Loss (DTL), with a possibility of the symbiont evolving temporarily outside the host phylogeny (in a “ghost” host lineage). The gene tree evolves similarly inside the symbiont tree, but horizontal transfers are constrained to symbionts co-occurring within the same host. Joint reconciliation scenarios are reconstructed and DTL event rates and likelihoods are estimated according to the model. As a nice addition, the authors propose a method to infer the symbiont phylogeny through amalgamation from gene trees and a host tree. The authors then explore the diverse possibilities offered by this method by testing it on both simulated datasets and biological datasets in order to check whether considering three nested levels is worthwhile. They convincingly show that three-level reconciliation has a better capacity to retrieve the symbiont donors and receivers of horizontal gene transfers, probably because transfers are constrained by additional elements relevant to the biological systems. Using, aphids, their obligate endosymbionts, and the symbiont genes involved in their nutritional functions, they identify horizontal gene transfers between aphid symbionts that are missed by two-level reconciliations but detected by expertise (Manzano-Marín et al. 2020). The other dataset presented here is on the human pathogen Helicobacter pylori, which history is supposed to reflect human migration. They use more than 1000 H. pylori gene families, and four populations, and use likelihood computations to compare different hypotheses on the diversification of the host. In summary, this study is a proof-of-concept of a 3-level reconciliation, where the authors manage to convey the applicability of their framework to many biological systems. Reported complexities, confirmed by reported running times, show that the method is computationally efficient. Without a doubt, the tool presented here will be very useful to evolutionary biologists who want to investigate multi-scale cophylogenies and it will move forward the study of associations between host and symbionts when symbiont genomic data are available. REFERENCES Lerat, E., Daubin, V., & Moran, N. A. (2003). From gene trees to organismal phylogeny in prokaryotes: the case of the γ-Proteobacteria. PLoS biology, 1(1), e19. Menet H, Trung AN, Daubin V, Tannier E (2023) Host-symbiont-gene phylogenetic reconciliation. bioRxiv, 2022.07.01.498457, ver. 2 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.07.01.498457 Szöllősi, G. J., Rosikiewicz, W., Boussau, B., Tannier, E., & Daubin, V. (2013). Efficient exploration of the space of reconciled gene trees. Systematic biology, 62(6), 901-912. | Host-symbiont-gene phylogenetic reconciliation | Hugo Menet, Alexia Nguyen Trung, Vincent Daubin, Eric Tannier | <p style="text-align: justify;"><strong>Motivation:</strong> Biological systems are made of entities organized at different scales e.g. macro-organisms, symbionts, genes...) which evolve in interaction.<br>These interactions range from indepe... | | Bioinformatics & Computational Biology, Phylogenetics / Phylogenomics | Emmanuelle Jousselin | 2022-08-21 18:34:27 | ||
02 Nov 2022
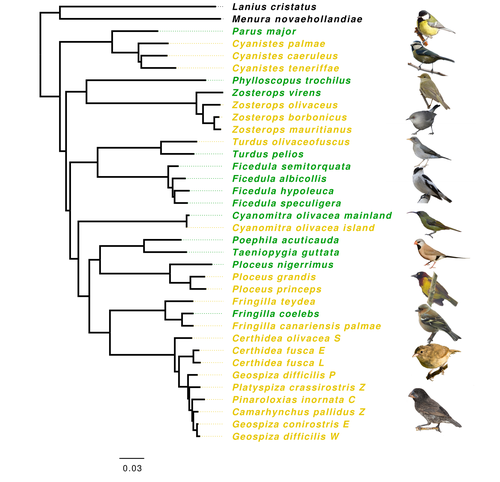
Evolution of immune genes in island birds: reduction in population sizes can explain island syndromeDemographic effects may affect adaptation to islandsRecommended by Emma Berdan based on reviews by Steven Fiddaman and 3 anonymous reviewersThe unique challenges associated with living on an island often result in organisms displaying a specific suite of traits commonly referred to as “island syndrome” (Adler and Levins, 1994; Burns, 2019; Baeckens and Van Damme, 2020). Large phenotypic shifts such as changes in size (e.g. shifts to gigantism or dwarfism, Lomolino, 2005) or coloration (Doutrelant et al., 2016) abound in the literature. However, less obvious phenotypes may also play a key role in adaptation to islands. One such trait, reduced immune function, has important implications for the future of island populations in the face of anthropogenic-induced changes. Due to lower parasite pressure caused by a less diverse and less virulent parasite population, island hosts may show a decrease in immune defenses (Beadell et al., 2006; Pérez‐Rodríguez et al., 2013). However, this hypothesis has been challenged, as many studies have found ambiguous or conflicting results (Matson, 2006; Illera et al., 2015). While most previous work has examined various immunological parameters (e.g., antibody concentrations), here, Barthe et al. (2022) take the novel approach of examining molecular signatures of immune genes. Using comparative genomic data from 34 different species of birds the authors examine the ratio of synonymous substitutions (i.e., not changing an amino acid) to non-synonymous substitutions (i.e., changing an amino acid) in innate and acquired immune genes (Pn/Ps ratio). Because population sizes on islands are lower which will affect molecular evolution, they compare these results to data from 97 control genes. Assuming relaxed selection on islands predicts that the difference between the Pn/Ps ratio of immune genes and of control genes (ΔPn/Ps) is greater in island species compared to mainland ones. As with previous work the authors found that the results differ depending on the category of immune genes. Both forms of innate defense: beta-defensins and Toll-like receptors did not show higher ΔPn/Ps for island populations. As these genes still have a higher Pn/Ps than control genes, the authors argue these results are in line with these genes being under purifying selection but lacking an “island effect”. Instead, the authors argue that demographic effects (i.e., reductions in Ne) may lead to the decreased immunity documented in other studies. In contrast, there was a reduction in Pn/Ps in MHC II genes, known to be under balancing selection. This reduction was stronger in island species and thus the authors argue that this is the only class of genes where a role for relaxed selection can be invoked. Together these results demonstrate that the changes in immunity experienced by island species are complex and that different categories of immune genes can experience different selective pressures. By including control genes in their study, they particularly highlight the importance of accounting for shifts in Ne when examining patterns of island species evolution. Hopefully, this kind of framework will be applied to other taxa to determine if these results are widespread or more specific to birds. References Adler GH, Levins R (1994) The Island Syndrome in Rodent Populations. The Quarterly Review of Biology, 69, 473–490. https://doi.org/10.1086/418744 Baeckens S, Van Damme R (2020) The island syndrome. Current Biology, 30, R338–R339. https://doi.org/10.1016/j.cub.2020.03.029 Barthe M, Doutrelant C, Covas R, Melo M, Illera JC, Tilak M-K, Colombier C, Leroy T, Loiseau C, Nabholz B (2022) Evolution of immune genes in island birds: reduction in population sizes can explain island syndrome. bioRxiv, 2021.11.21.469450, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2021.11.21.469450 Beadell JS, Ishtiaq F, Covas R, Melo M, Warren BH, Atkinson CT, Bensch S, Graves GR, Jhala YV, Peirce MA, Rahmani AR, Fonseca DM, Fleischer RC (2006) Global phylogeographic limits of Hawaii’s avian malaria. Proceedings of the Royal Society B: Biological Sciences, 273, 2935–2944. https://doi.org/10.1098/rspb.2006.3671 Burns KC (2019) Evolution in Isolation: The Search for an Island Syndrome in Plants. Cambridge University Press, Cambridge. https://doi.org/10.1017/9781108379953 Doutrelant C, Paquet M, Renoult JP, Grégoire A, Crochet P-A, Covas R (2016) Worldwide patterns of bird colouration on islands. Ecology Letters, 19, 537–545. https://doi.org/10.1111/ele.12588 Illera JC, Fernández-Álvarez Á, Hernández-Flores CN, Foronda P (2015) Unforeseen biogeographical patterns in a multiple parasite system in Macaronesia. Journal of Biogeography, 42, 1858–1870. https://doi.org/10.1111/jbi.12548 Lomolino MV (2005) Body size evolution in insular vertebrates: generality of the island rule. Journal of Biogeography, 32, 1683–1699. https://doi.org/10.1111/j.1365-2699.2005.01314.x Matson KD (2006) Are there differences in immune function between continental and insular birds? Proceedings of the Royal Society B: Biological Sciences, 273, 2267–2274. https://doi.org/10.1098/rspb.2006.3590 Pérez-Rodríguez A, Ramírez Á, Richardson DS, Pérez-Tris J (2013) Evolution of parasite island syndromes without long-term host population isolation: parasite dynamics in Macaronesian blackcaps Sylvia atricapilla. Global Ecology and Biogeography, 22, 1272–1281. https://doi.org/10.1111/geb.12084 | Evolution of immune genes in island birds: reduction in population sizes can explain island syndrome | Mathilde BARTHE, Claire DOUTRELANT, Rita COVAS, Martim MELO, Juan Carlos ILLERA, Marie-Ka TILAK, Constance COLOMBIER, Thibault LEROY , Claire LOISEAU , Benoit NABHOLZ | <p style="text-align: justify;">Shared ecological conditions encountered by species that colonize islands often lead to the evolution of convergent phenotypes, commonly referred to as “island syndrome”. Reduced immune functions have been previousl... | | Adaptation, Molecular Evolution, Population Genetics / Genomics | Emma Berdan | 2021-11-28 11:01:31 | ||
30 May 2023
slendr: a framework for spatio-temporal population genomic simulations on geographic landscapesA new powerful tool to easily encode the geo-spatial dimension in population genetics simulationsRecommended by Emiliano Trucchi based on reviews by Liisa Loog and 2 anonymous reviewers based on reviews by Liisa Loog and 2 anonymous reviewers
Models explaining the evolutionary processes operating in living beings are often impossible to test in the real world. This is mainly because of the long time (i.e., the number of generations) which is necessary for evolution to unfold. In addition, any such experiment would require a large number of individuals and, more importantly, many replicates to account for the inherent variance of the evolutionary processes under investigation. Only organisms with fast generation times and favourable rearing conditions can be used to explicitly test for specific evolutionary hypotheses. Computer simulations have filled this gap, revolutionising experimental testing in evolutionary biology by integrating genetic models into complex population dynamics, which can be run for (potentially) any length of time. Without going into an extensive description of the many available approaches for population genetics simulations (an exhaustive review can be found in Hoban et al 2012), three main aspects are, in my opinion, important for categorising and choosing one simulation approach over another. The first concerns the basic distinction between coalescent-based and individual-based simulators: the former being an efficient approach, which simulates back in time the coalescence events of a sample of homologous DNA fragments, while the latter is a more computationally intensive approach where all of the individuals (and their underlying genetic/genomic features) in the population are simulated forward-in-time, generation after generation. The second aspect concerns the simulation of natural selection. Although natural selection can be integrated into backward-in-time simulations, it is more realistically implemented as individual-based fitness in forward-in-time simulators. The third point, which has been often overlooked in evolutionary simulations, is about the possibility to design a simulation scenario where individuals and populations can exploit a physical (geographical) space. Amongst the coalescent-based simulators, SPLATCHE (Currat et al 2004), and its derivatives, is one of the few simulation tools deploying the coalescence process in sub-demes which are all connected by migration, thus getting as close as possible to a spatially-explicit population. On the other hand, individual-based simulators, whose development followed the increasing power of computational machines, offer a great opportunity to include spatio-temporal dynamics within a genomic simulation model. One of the most realistic and efficient individual-based forward-in-time simulators available is SLiM (Haller and Messer 2017), which allows users to implement simulations in arbitrarily complex spaces. Here, the more challenging part is encoding the spatially-explicit scenarios using the SLiM-specific EIDOS language. The new R package slendr (Petr et al 2022) offers a practical solution to this issue. By wrapping different tools into a well-known scripting language, slendr allows the design of spatiotemporal simulation scenarios which can be directly executed in the individual-based SLiM simulator, and the output stored with modern tree-sequence analysis tools (tskit; Kellerer et al 2018). Alternatively, simulations of non-spatial models can be run using a coalescent-based algorithm (msprime; Baumdicker et al 2022). The main advantage of slendr is that the whole simulative experiment can be performed entirely in the R environment, taking advantage of the many libraries available for geospatial and genomic data analysis, statistics, and visualisation. The open-source nature of this package, whose main aim is to make complex population genomics modelling more accessible, and the vibrant community of SLiM and tskit users will very likely make slendr widely used amongst the molecular ecology and evolutionary biology communities. Slendr handles real Earth cartographic data where users can design realistic demographic processes which characterise natural populations (i.e., expansions, displacement of large populations, interactions among populations, migrations, population splits, etc.) by changing spatial population boundaries across time and space. All in all, slendr is a very flexible and scalable framework to test the accuracy of spatial models, hypotheses about demography and selection, and interactions between organisms across space and time. REFERENCES Baumdicker, F., Bisschop, G., Goldstein, D., Gower, G., Ragsdale, A. P., Tsambos, G., ... & Kelleher, J. (2022). Efficient ancestry and mutation simulation with msprime 1.0. Genetics, 220(3), iyab229. https://doi.org/10.1093/genetics/iyab229 Currat, M., Ray, N., & Excoffier, L. (2004). SPLATCHE: a program to simulate genetic diversity taking into account environmental heterogeneity. Molecular Ecology Notes, 4(1), 139-142. https://doi.org/10.1046/j.1471-8286.2003.00582.x Haller, B. C., & Messer, P. W. (2017). SLiM 2: flexible, interactive forward genetic simulations. Molecular biology and evolution, 34(1), 230-240. https://doi.org/10.1093/molbev/msw211 Hoban, S., Bertorelle, G., & Gaggiotti, O. E. (2012). Computer simulations: tools for population and evolutionary genetics. Nature Reviews Genetics, 13(2), 110-122. https://doi.org/10.1038/nrg3130 Kelleher, J., Thornton, K. R., Ashander, J., & Ralph, P. L. (2018). Efficient pedigree recording for fast population genetics simulation. PLoS computational biology, 14(11), e1006581. https://doi.org/10.1371/journal.pcbi.1006581 Petr, M., Haller, B. C., Ralph, P. L., & Racimo, F. (2023). slendr: a framework for spatio-temporal population genomic simulations on geographic landscapes. bioRxiv, 2022.03.20.485041, ver. 5 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2022.03.20.485041 | slendr: a framework for spatio-temporal population genomic simulations on geographic landscapes | Martin Petr, Benjamin C. Haller, Peter L. Ralph, Fernando Racimo | <p style="text-align: justify;">One of the goals of population genetics is to understand how evolutionary forces shape patterns of genetic variation over time. However, because populations evolve across both time and space, most evolutionary proce... | | Bioinformatics & Computational Biology, Evolutionary Theory, Phylogeography & Biogeography, Population Genetics / Genomics | Emiliano Trucchi | 2022-09-14 12:57:56 | ||
02 Nov 2020

Experimental evolution of virulence and associated traits in a Drosophila melanogaster – Wolbachia symbiosisTemperature effects on virulence evolution of wMelPop Wolbachia in Drosophila melanogasterRecommended by Ellen Decaestecker based on reviews by Shira Houwenhuyse and 3 anonymous reviewersMonnin et al. [1] here studied how Drosophila populations are affected when exposed to a high virulent endosymbiotic wMelPop Wolbachia strain and why virulent vertically transmitting endosymbionts persist in nature. This virulent wMelPop strain has been described to be a blocker of dengue and other arboviral infections in arthropod vector species, such as Aedes aegypti. Whereas it can thus function as a mutualistic symbiont, it here acts as an antagonist along the mutualism-antagonism continuum symbionts operate. The wMelPop strain is not a natural occurring strain in Drosophila melanogaster and thus the start of this experiment can be seen as a novel host-pathogen association. Through experimental evolution of 17 generations, the authors studied how high temperature affects wMelPop Wolbachia virulence and Drosophila melanogaster survival. The authors used Drosophila strains that were selected for late reproduction, given that this should favor evolution to a lower virulence. Assumptions for this hypothesis are not given in the manuscript here, but it can indeed be assumed that energy that is assimilated to symbiont tolerance instead of reproduction may lead to reduced virulence evolution. This has equally been suggested by Reyserhove et al. [2] in a dynamics energy budget model tailored to Daphnia magna virulence evolution upon a viral infection causing White fat Cell disease, reconstructing changing environments through time. References [1] Monnin, D., Kremer, N., Michaud, C., Villa, M., Henri, H., Desouhant, E. and Vavre, F. (2020) Experimental evolution of virulence and associated traits in a Drosophila melanogaster – Wolbachia symbiosis. bioRxiv, 2020.04.26.062265, ver. 4 peer-reviewed and recommended by PCI Evol Biol. doi: https://doi.org/10.1101/2020.04.26.062265 | Experimental evolution of virulence and associated traits in a Drosophila melanogaster – Wolbachia symbiosis | David Monnin, Natacha Kremer, Caroline Michaud, Manon Villa, Hélène Henri, Emmanuel Desouhant, Fabrice Vavre | <p>Evolutionary theory predicts that vertically transmitted symbionts are selected for low virulence, as their fitness is directly correlated to that of their host. In contrast with this prediction, the Wolbachia strain wMelPop drastically reduces... | | Evolutionary Ecology, Experimental Evolution, Species interactions | Ellen Decaestecker | 2020-04-29 19:16:56 | ||
06 Oct 2022
Evolution of sperm morphology in a crustacean genus with fertilization inside an open brood pouch.Evolution of sperm morphology in Daphnia within a phyologenetic contextRecommended by Ellen Decaestecker based on reviews by Renate Matzke-Karasz and 1 anonymous reviewerIn this study sperm morphology is studied in 15 Daphnia species and the morphological data are mapped on a Daphnia phylogeny. The authors found that despite the internal fertilization mode, Daphnia have among the smallest sperm recorded, as would be expected with external fertilization. The authors also conclude that increase in sperm length has evolved twice, that sperm encapsulation has been lost in a clade, and that this clade has very polymorphic sperm with long, and often numerous, filopodia. Daphnia is an interesting model to study sperm morphology because the biology of sexual reproduction is often ignored in (cyclical) parthenogenetic species. Daphnia is part of the very diverse and successful group of cladocerans with cyclical parthenogenetic reproduction. The success of this reproduction mode is reflected in the known 620 species that radiated within this order, this is more than half of the known Branchiopod species diversity and the estimated number of cladoceran species is even two to four times higher (Forró et al. 2008). Looking at this particular model with a good phylogeny and some particularity in the mode of fertilization/reproduction, has thus a large value. Most Daphnia species are cyclical parthenogenetic and switch between sexual and asexual reproduction depending on the environmental conditions. Within the genus Daphnia, evolution to obligate asexuality has evolved in at least four independent occasions by three different mechanisms: (i) obligate parthenogenesis through hybridisation with or without polyploidy, (ii) asexuality has been acquired de novo in some populations and (iii) in certain lineages females reproduce by obligate parthenogenesis, whereas the clonally propagated males produce functional haploid sperm that allows them to breed with sexual females of normal cyclically parthenogenetic lineages (more on this in Decaestecker et al. 2009). This study is made in the context of a body of research on the evolution of one of the most fundamental and taxonomically diverse cell types. There is surprisingly little known about the adaptive value underlying their morphology because it is very difficult to test this experimentally. Studying sperm morphology across species is interesting to study evolution itself because it is a "simple trait". As the authors state: The understanding of the adaptive value of sperm morphology, such as length and shape, remains largely incomplete (Lüpold & Pitnick, 2018). Based on phylogenetic analyses across the animal kingdom, the general rule seems to be that fertilization mode (i.e. whether eggs are fertilized within or outside the female) is a key predictor of sperm length (Kahrl et al., 2021). There is a trade-off between sperm number and length (Immler et al., 2011). This study reports on one of the smallest sperm recorded despite the fertilization being internal. The brood pouch in Daphnia is an interesting particularity as fertilisation occurs internally, but it is not disconnected from the environment. It is also remarkable that there are two independent evolution lines of sperm size in this group. It suggests that those traits have an adaptive value. References Decaestecker E, De Meester L, Mergeay J (2009) Cyclical Parthenogenesis in Daphnia: Sexual Versus Asexual Reproduction. In: Lost Sex: The Evolutionary Biology of Parthenogenesis (eds Schön I, Martens K, Dijk P), pp. 295–316. Springer Netherlands, Dordrecht. https://doi.org/10.1007/978-90-481-2770-2_15 Duneau David, Möst M, Ebert D (2022) Evolution of sperm morphology in a crustacean genus with fertilization inside an open brood pouch. bioRxiv, 2020.01.31.929414, ver. 4 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.01.31.929414 Forró L, Korovchinsky NM, Kotov AA, Petrusek A (2008) Global diversity of cladocerans (Cladocera; Crustacea) in freshwater. Hydrobiologia, 595, 177–184. https://doi.org/10.1007/s10750-007-9013-5 Immler S, Pitnick S, Parker GA, Durrant KL, Lüpold S, Calhim S, Birkhead TR (2011) Resolving variation in the reproductive tradeoff between sperm size and number. Proceedings of the National Academy of Sciences, 108, 5325–5330. https://doi.org/10.1073/pnas.1009059108 Kahrl AF, Snook RR, Fitzpatrick JL (2021) Fertilization mode drives sperm length evolution across the animal tree of life. Nature Ecology & Evolution, 5, 1153–1164. https://doi.org/10.1038/s41559-021-01488-y Lüpold S, Pitnick S (2018) Sperm form and function: what do we know about the role of sexual selection? Reproduction, 155, R229–R243. https://doi.org/10.1530/REP-17-0536 | Evolution of sperm morphology in a crustacean genus with fertilization inside an open brood pouch. | Duneau, David; Moest, Markus; Ebert, Dieter | <p style="text-align: justify;">Sperm is the most fundamental male reproductive feature. It serves the fertilization of eggs and evolves under sexual selection. Two components of sperm are of particular interest, their number and their morphology.... | | Evolutionary Ecology, Morphological Evolution, Reproduction and Sex, Sexual Selection | Ellen Decaestecker | 2020-05-30 22:54:15 | ||
14 Apr 2021

Parasitic success and venom composition evolve upon specialization of parasitoid wasps to different host speciesWhat makes a parasite successful? Parasitoid wasp venoms evolve rapidly in a host-specific mannerRecommended by Élio Sucena based on reviews by Simon Fellous, alexandre leitão and 1 anonymous reviewerParasitoid wasps have developed different mechanisms to increase their parasitic success, usually at the expense of host survival (Fellowes and Godfray, 2000). Eggs of these insects are deposited inside the juvenile stages of their hosts, which in turn deploy several immune response strategies to eliminate or disable them (Yang et al., 2020). Drosophila melanogaster protects itself against parasitoid attacks through the production of specific elongated haemocytes called lamellocytes which form a capsule around the invading parasite (Lavine and Strand, 2002; Rizki and Rizki, 1992) and the subsequent activation of the phenol-oxidase cascade leading to the release of toxic radicals (Nappi et al., 1995). On the parasitoid side, robust responses have evolved to evade host immune defenses as for example the Drosophila-specific endoparasite Leptopilina boulardi, which releases venom during oviposition that modifies host behaviour (Varaldi et al., 2006) and inhibits encapsulation (Gueguen et al., 2011; Martinez et al., 2012).
References Cavigliasso, F., Mathé-Hubert, H., Gatti, J.-L., Colinet, D. and Poirié, M. (2021) Parasitic success and venom composition evolve upon specialization of parasitoid wasps to different host species. bioRxiv, 2020.10.24.353417, ver. 3 peer-reviewed and recommended by Peer Community in Evolutionary Biology. https://doi.org/10.1101/2020.10.24.353417 Cavigliasso, F., Mathé-Hubert, H., Kremmer, L., Rebuf, C., Gatti, J.-L., Malausa, T., Colinet, D., Poiré, M. and Léne. (2019). Rapid and Differential Evolution of the Venom Composition of a Parasitoid Wasp Depending on the Host Strain. Toxins, 11(629). https://doi.org/10.3390/toxins11110629 Colinet, D., Deleury, E., Anselme, C., Cazes, D., Poulain, J., Azema-Dossat, C., Belghazi, M., Gatti, J. L. and Poirié, M. (2013). Extensive inter- and intraspecific venom variation in closely related parasites targeting the same host: The case of Leptopilina parasitoids of Drosophila. Insect Biochemistry and Molecular Biology, 43(7), 601–611. https://doi.org/10.1016/j.ibmb.2013.03.010 Colinet, D., Dubuffet, A., Cazes, D., Moreau, S., Drezen, J. M. and Poirié, M. (2009). A serpin from the parasitoid wasp Leptopilina boulardi targets the Drosophila phenoloxidase cascade. Developmental and Comparative Immunology, 33(5), 681–689. https://doi.org/10.1016/j.dci.2008.11.013 Fellowes, M. D. E. and Godfray, H. C. J. (2000). The evolutionary ecology of resistance to parasitoids by Drosophila. Heredity, 84(1), 1–8. https://doi.org/10.1046/j.1365-2540.2000.00685.x Gueguen, G., Rajwani, R., Paddibhatla, I., Morales, J. and Govind, S. (2011). VLPs of Leptopilina boulardi share biogenesis and overall stellate morphology with VLPs of the heterotoma clade. Virus Research, 160(1–2), 159–165. https://doi.org/10.1016/j.virusres.2011.06.005 Lavine, M. D. and Strand, M. R. (2002). Insect hemocytes and their role in immunity. Insect Biochemistry and Molecular Biology, 32(10), 1295–1309. https://doi.org/10.1016/S0965-1748(02)00092-9 Martinez, J., Duplouy, A., Woolfit, M., Vavre, F., O’Neill, S. L. and Varaldi, J. (2012). Influence of the virus LbFV and of Wolbachia in a host-parasitoid interaction. PloS One, 7(4), e35081. https://doi.org/10.1371/journal.pone.0035081 Nappi, A. J., Vass, E., Frey, F. and Carton, Y. (1995). Superoxide anion generation in Drosophila during melanotic encapsulation of parasites. European Journal of Cell Biology, 68(4), 450–456. Poirié, M., Colinet, D. and Gatti, J. L. (2014). Insights into function and evolution of parasitoid wasp venoms. Current Opinion in Insect Science, 6, 52–60. https://doi.org/10.1016/j.cois.2014.10.004 Rizki, T. M. and Rizki, R. M. (1992). Lamellocyte differentiation in Drosophila larvae parasitized by Leptopilina. Developmental and Comparative Immunology, 16(2–3), 103–110. https://doi.org/10.1016/0145-305X(92)90011-Z Schlenke, T. A., Morales, J., Govind, S. and Clark, A. G. (2007). Contrasting infection strategies in generalist and specialist wasp parasitoids of Drosophila melanogaster. PLoS Pathogens, 3(10), 1486–1501. https://doi.org/10.1371/journal.ppat.0030158 Varaldi, J., Petit, S., Boulétreau, M. and Fleury, F. (2006). The virus infecting the parasitoid Leptopilina boulardi exerts a specific action on superparasitism behaviour. Parasitology, 132(Pt 6), 747–756. https://doi.org/10.1017/S0031182006009930 Yang, L., Qiu, L., Fang, Q., Stanley, D. W. and Gong‐Yin, Y. (2020). Cellular and humoral immune interactions between Drosophila and its parasitoids. Insect Science. https://doi.org/10.1111/1744-7917.12863
| Parasitic success and venom composition evolve upon specialization of parasitoid wasps to different host species | Fanny Cavigliasso, Hugo Mathé-Hubert, Jean-Luc Gatti, Dominique Colinet, Marylène Poirié | <p>Female endoparasitoid wasps usually inject venom into hosts to suppress their immune response and ensure offspring development. However, the parasitoid’s ability to evolve towards increased success on a given host simultaneously with the evolut... | | Experimental Evolution, Species interactions | Élio Sucena | 2020-10-26 15:00:55 | ||
05 May 2020
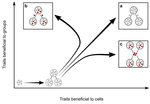
Meta-population structure and the evolutionary transition to multicellularityThe ecology of evolutionary transitions to multicellularityRecommended by Dustin Brisson based on reviews by 2 anonymous reviewersThe evolutionary transition to multicellular life from free-living, single-celled ancestors has occurred independently in multiple lineages [1-5]. This evolutionary transition to cooperative group living can be difficult to explain given the fitness advantages enjoyed by the non-cooperative, single-celled organisms that still numerically dominate life on earth [1,6,7]. Although several hypotheses have been proposed to explain the transition to multicellularity, a common theme is the abatement of the efficacy of natural selection among the single cells during the free-living stage and the promotion of the efficacy of selection among groups of cells during the cooperative stage, an argument reminiscent of those from George Williams’ seminal book [8,9]. The evolution of life cycles appears to be a key step in the transition to multicellularity as it can align fitness advantages of the single-celled 'reproductive' stage with that of the cooperative 'organismal' stage [9-12]. That is, the evolution of life cycles allows natural selection to operate over timescales longer than that of the doubling time of the free-living cells [13]. Despite the importance of this issue, identifying the range of ecological conditions that reduce the importance of natural selection at the single-celled, free-living stage and increase the importance of selection among groups of cooperating cells has not been addressed empirically. References [1] Maynard Smith, J. and Szathmáry, E. (1995). The Major Transitions in Evolution. Oxford, UK: Freeman. p 346. | Meta-population structure and the evolutionary transition to multicellularity | Caroline J Rose, Katrin Hammerschmidt, Yuriy Pichugin and Paul B Rainey | <p>The evolutionary transition to multicellularity has occurred on numerous occasions, but transitions to complex life forms are rare. While the reasons are unclear, relevant factors include the intensity of within- versus between-group selection ... | | Adaptation, Evolutionary Dynamics, Experimental Evolution | Dustin Brisson | 2019-04-04 12:26:36 |




