Thermal regimes, but not mean temperatures, drive patterns of rapid climate adaptation at a continent-scale: evidence from the introduced European earwig across North America
Jean-Claude Tourneur, Joël Meunier
https://doi.org/10.1101/550319
Temperature variance, rather than mean, drives adaptation to local climate
Recommended by Fabien Aubret based on reviews by Ben Phillips and Eric Gangloff
Climate change is impacting eco-systems worldwide and driving many populations to move, adapt or go extinct. It is increasingly appreciated, for example, that species may adjust their phenology in response to climate change, although empirical data is scarce. In this preprint [1], Tourneur and Meunier report an impressive sampling effort in which life-history traits were measured across introduced populations of earwig in North America. The authors examine whether variation in life-history across populations is correlated with aspects of the thermal climate experienced by each population: mean temperature and seasonality of temperature. They find some fascinating correlations between life-history and thermal climate; correlations with the seasonality of temperature, but not with mean temperature. This study provides relatively uncommon data, in the sense that where most of the literature looking at adaptation in animals in response to climate change has focused on physiological traits [2, 3], this study examines changes in life-history traits with time scales relevant to impending climate change, and provides a reasonable argument that this is adaptation, not just constraint.
References
[1] Tourneur, J.-C. and Meunier, J. (2019). Thermal regimes, but not mean temperatures, drive patterns of rapid climate adaptation at a continent-scale: evidence from the introduced European earwig across North America. BioRxiv, 550319, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/550319
[2] Kellermann, V., Overgaard, J., Hoffmann, A. A., Fløjgaard, C., Svenning, J. C., & Loeschcke, V. (2012). Upper thermal limits of Drosophila are linked to species distributions and strongly constrained phylogenetically. Proceedings of the National Academy of Sciences, 109(40), 16228-16233. doi: 10.1073/pnas.1207553109
[3] Hoffmann, A. A., & Sgro, C. M. (2011). Climate change and evolutionary adaptation. Nature, 470(7335), 479. doi: 10.1038/nature09670
| Thermal regimes, but not mean temperatures, drive patterns of rapid climate adaptation at a continent-scale: evidence from the introduced European earwig across North America | Jean-Claude Tourneur, Joël Meunier | <p>The recent development of human societies has led to major, rapid and often inexorable changes in the environment of most animal species. Over the last decades, a growing number of studies formulated predictions on the modalities of animal adap... |  | Adaptation, Evolutionary Ecology, Life History | Fabien Aubret | | 2019-02-15 09:12:11 | View |
Environmental specificity in Drosophila-bacteria symbiosis affects host developmental plasticity
Robin Guilhot, Antoine Rombaut, Anne Xuéreb, Kate Howell, Simon Fellous
https://doi.org/10.1101/717702
Nutrition-dependent effects of gut bacteria on growth plasticity in Drosophila melanogaster
Recommended by Wolf Blanckenhorn based on reviews by Pedro Simões and 1 anonymous reviewer
It is well known that the rearing environment has strong effects on life history and fitness traits of organisms. Microbes are part of every environment and as such likely contribute to such environmental effects. Gut bacteria are a special type of microbe that most animals harbor, and as such they are part of most animals’ environment. Such microbial symbionts therefore likely contribute to local adaptation [1]. The main question underlying the laboratory study by Guilhot et al. [2] was: How much do particular gut bacteria affect the organismal phenotype, in terms of life history and larval foraging traits, of the fruit fly Drosophila melanogaster, a common laboratory model species in biology?
To investigate the above question, the authors isolated 4 taxa of bacteria from the gut of a (randomly picked) Drosophila melanogaster lab strain, and subsequently let Drosophila melanogaster eggs and larvae (stemming from their own, different lab strain) develop both in the typical artificial laboratory medium as well as in grapes, a natural “new” habitat for Drosophila larvae, inoculated with theses bacteria, singly and in combination, also including a bacteria-free control. By investigating various relevant developmental and size traits, the authors found that adding particularly Enterobacteria had some visible effects on several traits, both upward (indicting improvement) and downward (being detrimental) (with three other types of bacteria showing only minor or even no effects). In general, the grape medium reduced performance relative to the standard lab medium. Strongest interactive effects occurred for development time and body size, together making up growth plasticity [3], with lesser such effects on some related behavioral (feeding) traits (Figs. 2,3).
The study premise is interesting, its general objectives are clearly laid out, and the practical work was conducted correctly as far as I can evaluate. The study remains largely descriptive in that no particular a priori hypotheses or predictions in relation to the specific bacteria isolated were formulated, not least because the bacteria were necessarily somewhat arbitrarily chosen and there were apparently no prior studies from which to derive concrete predictions. Overall, the results of this study should be of interest to the community of evolutionary ecologists, especially those working on nutritional and microbiome effects on animal life histories. I consider this work to be primarily ecological, with limited evolutionary content (e.g. no genetics) though some evolutionary implications, as mentioned in the paper’s Conclusions. So this paper would best fit in a microbial or physiological ecology outlet/journal.
The inclusion of a natural medium (grapes) must be commended because this permits inferences and conclusions for at least one natural environment, whereas inferences drawn from laboratory studies in the artificial medium that most Drosophila researchers seem to use are typically limited. Unsurprisingly perhaps, the study showed that Drosophila melanogaster fared generally better in the artificial than the chosen natural medium (grape). Crucially, however, the bacterial symbionts modified both media differentially. Although common bacterial taxa were chosen, the particular bacteria isolated and used remain arbitrary, as there are many. I note that the main and strongest interactive effects between medium and bacterial type are apparent for the Enterobacteria, and they probably also strongly, if not exclusively, mediate the overall effect of the bacterial mixture.
While these specific data are novel, they are not very surprising. If we grow animals in different environments we can expect some detectable effects of these environments, including the bacterial (microbiome) environment, on the hosts life history. The standard and predicted [4] life history response of Drosophila melanogaster (but not all insects [3]) facing stressful nutritional environments, as apparently created by the Enterobacteria, is to extend development but come out smaller in the end. This is what happened here for the laboratory medium ([2]: Fig. 5). The biological interpretation is that individuals have more trouble ingesting and/or digesting the nutrients available (thus prolonging their foraging period and development), yet cannot convert the nutrients effectively into body size increments (hence emerging smaller). This is what the authors here refer to as developmental plasticity, which is ultimately nutritionally mediated. However, interestingly, a signal in the opposite direction was indicated for the bacterial mixture in the grape medium (flies emerging larger after accelerated development: Fig. 5), suggesting some positive effects on growth rate of the natural medium, perhaps related to grapes being a limited resource that needs to be escaped quickly [3]? The reversal of sexual size dimorphism across bacterial treatments in the grape environment detectable in Fig. 4 is interesting, too, though I don’t understand why this happens, and this is not discussed.
In general, more encompassing and increased questions in this context to be researched in the future could be: 1) are these effects predictable (not (yet) at this point, or so it seems); and 2) how strong are these environmental bacterial effects relative to other, more standard effects (e.g. relative to genetic variation, population variation, etc., or relative to other types of environmental effects like, say, temperature)? (3) It could further be asked why not natural but laboratory populations of Drosophila were used for this experiment, if the aim was to draw inferences for the wild situation. (4) Although Genotype x Environment effects are invoked in the Discussion, they were not tested here, lacking genetically different Drosophila families or populations. From an evolutionary standpoint, I consider this the greatest weakness of the study. I was also not too thrilled by the particular statistical analyses employed, though this ultimately does not negate the results. Nevertheless, this work is a good start in this huge field investigating the microbiome. In conclusion, I can recommend this paper after review by PCI Evol Biol.
References
[1] Kawecki, T. J. and Ebert, D. (2004) Conceptual issues in local adaptation. Ecology Letters 7: 1225-1241. doi: 10.1111/j.1461-0248.2004.00684.x
[2] Guilhot, R., Rombaut, A., Xuéreb, A., Howell, K. and Fellous, S. (2019). Environmental specificity in Drosophila-bacteria symbiosis affects host developmental plasticity. BioRxiv, 717702, v3 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/717702
[3] Blanckenhorn, W.U. (1999) Different growth responses to temperature and resource limitation in three fly species with similar life histories. Evolutionary Ecology 13: 395-409. doi: 10.1023/A:1006741222586
[4] Stearns, S. C. and Koella, J. (1986) The evolution of phenotypic plasticity in life history traits: predictions of reaction norms for age and size at maturity. Evolution 40: 893-914. doi: 10.1111/j.1558-5646.1986.tb00560.x
| Environmental specificity in Drosophila-bacteria symbiosis affects host developmental plasticity | Robin Guilhot, Antoine Rombaut, Anne Xuéreb, Kate Howell, Simon Fellous | <p>Environmentally acquired microbial symbionts could contribute to host adaptation to local conditions like vertically transmitted symbionts do. This scenario necessitates symbionts to have different effects in different environments. We investig... |  | Adaptation, Evolutionary Ecology, Phenotypic Plasticity, Species interactions | Wolf Blanckenhorn | | 2019-02-13 15:22:23 | View |
Tell me who you mate with, I’ll tell you what’s going on
Recommended by Sara Magalhaes and Alexandre Courtiol based on reviews by Alexandre Courtiol and 2 anonymous reviewers
The study of sexual selection goes as far as Darwin himself. Since then, elaborate theories concerning both intra- and inter-sexual sexual have been developed, and elegant experiments have been designed to test this body of theory. It may thus come as a surprise that the community is still debating on the correct way to measure simple components of sexual selection, such as the Bateman gradient (i.e., the covariance between the number of matings and the number of offspring)[1,2], or to quantify complex behaviours such as mate choice (the non-random choice of individuals with particular characters as mates)[3,4] and their consequences.
One difficulty in the study of sexual selection is evaluating the consequences of non-random mating. Indeed, when non-random mating is observed in a population, it is often difficult to establish whether such mating pattern leads to i) sexual selection per se (selection pressures favouring certain phenotypes), and/or ii) the non-random association of parental genes in their offspring or not. These two processes differ. In particular, assortative (and disassortative) mating can shape genetic covariances without leading to changes in gene frequencies in the population. Their distinction matters because these two processes lead to different evolutionary outcomes, which can have large ripple effects in the evolution of sexual behaviours, sexual ornamentation, and speciation.
In his paper, entitled “Multi-model inference of non-random mating from an information theoretic approach” [5], Carvajal-Rodríguez tackled this issue. The author generated a simple model in which the consequences of non-random mating can be inferred from information on the population frequencies before and after mating. The procedure is as follows: from the initial population frequencies of phenotypes (or genotypes) of both sexes, the model generates predictions on the frequencies after mating, assuming that particular mating patterns have occurred. This leads to different predictions for the phenotypic (or genotypic) frequencies after mating. The particular mating pattern leading to the best fit with the real frequencies is then identified via a model selection procedure (performing model averaging to combine different mating patterns is also possible).
This study builds on a framework introduced by Carvajal-Rodríguez’s colleagues [6] and encompasses later methodological developments involving the author himself [7]. Compared to early work, the new method proposed by the author builds on the relationship between mating pattern and information [8] to distinguish among scenarios that would lead to non-random mating due to different underlying processes, using simple model selection criterion such as the AICc.
The great asset of the proposed method is that it can be applied to the study of natural populations in which the study of mate choice and sexual selection is notoriously difficult. In the manuscript, the procedure is tested on a population of marine gastropods (Littorina saxatilis). This allows the reader to grasp how the method can be applied to a real system. In fact, anyone can try out the method thanks to the freely available software InfoMating programmed by the author. One important assumption underlying the current method is that the frequencies of unmated individuals do not change during the mating season. If this is not the case, the reader may refer to another publication of the same author which relaxes this assumption [9]. These papers are both instrumental for empiricists interested in testing sexual selection theory.
References
[1] Bateman, A. J. (1948). Intra-sexual selection in Drosophila. Heredity, 2(3), 349-368. doi: 10.1038/hdy.1948.21
[2] Jones, A. G. (2009). On the opportunity for sexual selection, the Bateman gradient and the maximum intensity of sexual selection. Evolution: International Journal of Organic Evolution, 63(7), 1673-1684. doi: 10.1111/j.1558-5646.2009.00664.x
[3] Andersson, M., & Simmons, L. W. (2006). Sexual selection and mate choice. Trends in ecology & evolution, 21(6), 296-302. doi: 10.1016/j.tree.2006.03.015
[4] Kuijper, B., Pen, I., & Weissing, F. J. (2012). A guide to sexual selection theory. Annual Review of Ecology, Evolution, and Systematics, 43, 287-311. doi: 10.1146/annurev-ecolsys-110411-160245
[5] Carvajal-Rodríguez, A. (2019). Multi-model inference of non-random mating from an information theoretic approach. bioRxiv, 305730, ver. 5 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/305730
[6] Rolán‐Alvarez, E., & Caballero, A. (2000). Estimating sexual selection and sexual isolation effects from mating frequencies. Evolution, 54(1), 30-36. doi: 10.1111/j.0014-3820.2000.tb00004.x
[7] Carvajal-Rodríguez, A., & Rolan-Alvarez, E. (2006). JMATING: a software for the analysis of sexual selection and sexual isolation effects from mating frequency data. BMC Evolutionary Biology, 6(1), 40. doi: 10.1186/1471-2148-6-40
[8] Carvajal-Rodríguez, A. (2018). Non-random mating and information theory. Theoretical population biology, 120, 103-113. doi: 10.1016/j.tpb.2018.01.003
[9] Carvajal-Rodríguez, A. (2019). A generalization of the informational view of non-random mating: Models with variable population frequencies. Theoretical population biology, 125, 67-74. doi: 10.1016/j.tpb.2018.12.004
| Multi-model inference of non-random mating from an information theoretic approach | Antonio Carvajal-Rodríguez | <p>Non-random mating has a significant impact on the evolution of organisms. Here, I developed a modelling framework for discrete traits (with any number of phenotypes) to explore different models connecting the non-random mating causes (mate comp... | 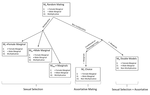 | Evolutionary Ecology, Evolutionary Theory, Sexual Selection | Sara Magalhaes | | 2019-02-08 19:24:03 | View |
Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates
Rylan Shearn, Alison E. Wright, Sylvain Mousset, Corinne Régis, Simon Penel, Jean-François Lemaitre, Guillaume Douay, Brigitte Crouau-Roy, Emilie Lecompte, Gabriel A.B. Marais
https://doi.org/10.1101/445072
Studying genetic antagonisms as drivers of genome evolution
Recommended by Mathieu Joron based on reviews by Qi Zhou and 3 anonymous reviewers
Sex chromosomes are special in the genome because they are often highly differentiated over much of their lengths and marked by degenerative evolution of their gene content. Understanding why sex chromosomes differentiate requires deciphering the forces driving their recombination patterns. Suppression of recombination may be subject to selection, notably because of functional effects of locking together variation at different traits, as well as longer-term consequences of the inefficient purge of deleterious mutations, both of which may contribute to patterns of differentiation [1]. As an example, male and female functions may reveal intrinsic antagonisms over the optimal genotypes at certain genes or certain combinations of interacting genes. As a result, selection may favour the recruitment of rearrangements blocking recombination and maintaining the association of sex-antagonistic allele combinations with the sex-determining locus.
The hypothesis that sexually antagonistic selection might drive recombination suppression along the sex chromosomes is not new, but there are surprisingly few studies examining this empirically [1]. Support mainly comes from the study of guppy populations Poecilia reticulata in which the level of sexual dimorphism (notably due to male ornaments, subject to sexual selection) varies among populations, and was found to correlate with the length of the non-recombining region on the sex chromosome [2]. But the link is not always that clear. For instance in the fungus Microbotryum violaceum, the mating type loci is characterized by adjacent segments with recombination suppression, despite the near absence of functional differentiation between mating types [3].
In this study, Shearn and colleagues [4] explore the patterns of recombination suppression on the sex chromosomes of primates. X and Y chromosomes are strongly differentiated, except in a small region where they recombine with each other, the pseudoautosomal region (PAR). In the clade of apes and monkeys, including humans, large rearrangements have extended the non recombining region stepwise, eroding the PAR. Could this be driven by sexually antagonistic selection in a clade showing strong sexual differentiation?
To evaluate this idea, Shearn et al. have compared the structure of recombination in apes and monkeys to their sister clade with lower levels of sexual dimorphism, the lemurs and the lorises. If sexual antagonism was important in shaping recombination suppression, and assuming lower measures of sexual dimorphism reflect lower sexual antagonism [5], then lemurs and lorises would be predicted to show a shorter non-recombining region than apes and monkeys.
Lemurs and lorises were terra incognita in terms of genomic research on the sex chromosomes, so Shearn et al. have sequenced the genomes of males and females of different species. To assess whether sequences came from a recombining or non-recombining segment, they used coverage information in males vs females to identify sequences on the X whose copy on the Y is absent or too divergent to map, indicating long-term differentiation (absence of recombination). This approach reveals that the two lineages have undergone different recombination dynamics since they split from their common ancestor: regions which have undergone further structural rearrangements extending the non-recombining region in apes and monkeys, have continued to recombine normally in lemurs and lorises. Consistent with the prediction, macroevolutionary variation in the differentiation of males and females is indeed accompanied by variation in the size of the non-recombining region on the sex chromosome.
Sex chromosomes are excellent examples of how genomes are shaped by selection. By directly exploring recombination patterns on the sex chromosome across all extant primate groups, this study comes as a nice addition to the short series of empirical studies evaluating whether sexual antagonism may drive certain aspects of genome structure. The sexual selection causing sometimes spectacular morphological or behavioural differences between sexes in many animals may be the visible tip of the iceberg of all the antagonisms that characterise male vs. female functions generally [5]. Further research should bring insight into how different flavours or intensities of antagonistic selection can contribute to shape genome variation.
References
[1] Charlesworth D (2017) Evolution of recombination rates between sex chromosomes. Philosophical Transactions of the Royal Society B: Biological Sciences, 372, 20160456. https://doi.org/10.1098/rstb.2016.0456
[2] Wright AE, Darolti I, Bloch NI, Oostra V, Sandkam B, Buechel SD, Kolm N, Breden F, Vicoso B, Mank JE (2017) Convergent recombination suppression suggests role of sexual selection in guppy sex chromosome formation. Nature Communications, 8, 14251. https://doi.org/10.1038/ncomms14251
[3] Branco S, Badouin H, Vega RCR de la, Gouzy J, Carpentier F, Aguileta G, Siguenza S, Brandenburg J-T, Coelho MA, Hood ME, Giraud T (2017) Evolutionary strata on young mating-type chromosomes despite the lack of sexual antagonism. Proceedings of the National Academy of Sciences, 114, 7067–7072. https://doi.org/10.1073/pnas.1701658114
[4] Shearn R, Wright AE, Mousset S, Régis C, Penel S, Lemaitre J-F, Douay G, Crouau-Roy B, Lecompte E, Marais GAB (2020) Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates. bioRxiv, 445072. https://doi.org/10.1101/445072
[5] Connallon T, Clark AG (2014) Evolutionary inevitability of sexual antagonism. Proceedings of the Royal Society B: Biological Sciences, 281, 20132123. https://doi.org/10.1098/rspb.2013.2123
| Evolutionary stasis of the pseudoautosomal boundary in strepsirrhine primates | Rylan Shearn, Alison E. Wright, Sylvain Mousset, Corinne Régis, Simon Penel, Jean-François Lemaitre, Guillaume Douay, Brigitte Crouau-Roy, Emilie Lecompte, Gabriel A.B. Marais | <p>Sex chromosomes are typically comprised of a non-recombining region and a recombining pseudoautosomal region. Accurately quantifying the relative size of these regions is critical for sex chromosome biology both from a functional (i.e. number o... | 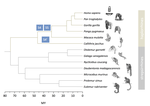 | Bioinformatics & Computational Biology, Genome Evolution, Molecular Evolution, Reproduction and Sex, Sexual Selection | Mathieu Joron | | 2019-02-04 15:16:32 | View |
Fitting diversification models on undated or partially dated trees
Recommended by Nicolas Lartillot based on reviews by Amaury Lambert, Dominik Schrempf and 1 anonymous reviewer
Phylogenetic trees can be used to extract information about the process of diversification that has generated them. The most common approach to conduct this inference is to rely on a likelihood, defined here as the probability of generating a dated tree T given a diversification model (e.g. a birth-death model), and then use standard maximum likelihood. This idea has been explored extensively in the context of the so-called diversification studies, with many variants for the models and for the questions being asked (diversification rates shifting at certain time points or in the ancestors of particular subclades, trait-dependent diversification rates, etc).
However, all this assumes that the dated tree T is known without error. In practice, trees (that is, both the tree topology and the divergence times) are inferred based on DNA sequences, possibly combined with fossil information for calibrating and informing the divergence times. Molecular dating is a delicate exercise, however, and much more so in fact than reconstructing the tree topology. In particular, a mis-specificied model for the relaxed molecular clock, or a mis-specifiied prior, can have a substantial impact on the estimation of divergence dates - which in turn could severely mislead the inference about the underlying diversification process. This thus raises the following question: would that be possible to conduct inference and testing of diversification models without having to go through the dangerous step of molecular dating?
In his article ""Probabilities of tree topologies with temporal constraints and diversification shifts"" [1], Gilles Didier introduces a recursive method for computing the probability of a tree topology under some diversification model of interest, without knowledge of the exact dates, but only interval constraints on the dates of some of the nodes of the tree. Such interval constraints, which are derived from fossil knowledge, are typically used for molecular dating: they provide the calibrations for the relaxed clock analysis. Thus, what is essentially proposed by Gilles Didier is to use them in combination with the tree topology only, thus bypassing the need to estimates divergence times first, before fitting a diversification model to a phylogenetic tree.
This article, which is primarily a mathematical and algorithmic contribution, is then complemented with several applications: testing for a diversification shift in a given subclade of the phylogeny, just based on the (undated) tree topology, with interval constraints on some of its internal nodes; but also, computing the age distribution of each node and sampling on the joint distribution on node ages, conditional on the interval constraints. The test for the presence of a diversification shift is particularly interesting: an application to simulated data (and without any interval constraint in that case) suggests that the method based on the undated tree performs about as well as the classical method based on a dated tree, and this, even granting the classical approach a perfect knowledge of the dates - given that, in practice, one in fact relies on potentially biased estimates. Finally, an application to a well-known example (rate shifts in cetacean phylogeny) is presented.
This article thus represents a particularly meaningful contribution to the methodology for diversification studies; but also, for molecular dating itself: it is a well known problem in molecular dating that computing and sampling from the conditional distributions on node ages, given fossil constraints, and more generally understanding and visualizing how interval constraints on some nodes of the tree impact the distribution at other nodes, is a particularly difficult exercise. For that reason, the algorithmic routines presented in the present article will be useful in this context as well.
References
[1] Didier, G. (2020) Probabilities of tree topologies with temporal constraints and diversification shifts. bioRxiv, 376756, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/376756
| Probabilities of tree topologies with temporal constraints and diversification shifts | Gilles Didier | <p>Dating the tree of life is a task far more complicated than only determining the evolutionary relationships between species. It is therefore of interest to develop approaches apt to deal with undated phylogenetic trees. The main result of this ... |  | Bioinformatics & Computational Biology, Macroevolution | Nicolas Lartillot | | 2019-01-30 11:28:58 | View |
A bird’s white-eye view on neosex chromosome evolution
Thibault Leroy, Yoann Anselmetti, Marie-Ka Tilak, Sèverine Bérard, Laura Csukonyi, Maëva Gabrielli, Céline Scornavacca, Borja Milá, Christophe Thébaud, Benoit Nabholz
https://doi.org/10.1101/505610
Young sex chromosomes discovered in white-eye birds
Recommended by Kateryna Makova based on reviews by Gabriel Marais, Melissa Wilson and 1 anonymous reviewer
Recent advances in next-generation sequencing are allowing us to uncover the evolution of sex chromosomes in non-model organisms. This study [1] represents an example of this application to birds of two Sylvioidea species from the genus Zosterops (commonly known as white-eyes). The study is exemplary in the amount and types of data generated and in the thoroughness of the analysis applied. Both male and female genomes were sequenced to allow the authors to identify sex-chromosome specific scaffolds. These data were augmented by generating the transcriptome (RNA-seq) data set. The findings after the analysis of these extensive data are intriguing: neoZ and neoW chromosome scaffolds and their breakpoints were identified. Novel sex chromosome formation appears to be accompanied by translocation events. The timing of formation of novel sex chromosomes was identified using molecular dating and appears to be relatively recent. Yet first signatures of distinct evolutionary patterns of sex chromosomes vs. autosomes could be already identified. These include the accumulation of transposable elements and changes in GC content. The changes in GC content could be explained by biased gene conversion and altered recombination landscape of the neo sex chromosomes. The authors also study divergence and diversity of genes located on the neo sex chromosomes. Here their findings appear to be surprising and need further exploration. The neoW chromosome already shows unique patterns of divergence and diversity at protein-coding genes as compared with genes on either neoZ or autosomes. In contrast, the genes on the neoZ chromosome do not display divergence or diversity patterns different from those for autosomes. This last observation is puzzling and I believe should be explored in further studies. Overall, this study significantly advances our knowledge of the early stages of sex chromosome evolution in vertebrates, provides an example of how such a study could be conducted in other non-model organisms, and provides several avenues for future work.
References
[1] Leroy T., Anselmetti A., Tilak M.K., Bérard S., Csukonyi L., Gabrielli M., Scornavacca C., Milá B., Thébaud C. and Nabholz B. (2019). A bird’s white-eye view on neo-sex chromosome evolution. bioRxiv, 505610, ver. 4 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/505610
| A bird’s white-eye view on neosex chromosome evolution | Thibault Leroy, Yoann Anselmetti, Marie-Ka Tilak, Sèverine Bérard, Laura Csukonyi, Maëva Gabrielli, Céline Scornavacca, Borja Milá, Christophe Thébaud, Benoit Nabholz | <p>Chromosomal organization is relatively stable among avian species, especially with regards to sex chromosomes. Members of the large Sylvioidea clade however have a pair of neo-sex chromosomes which is unique to this clade and originate from a p... |  | Molecular Evolution, Population Genetics / Genomics | Kateryna Makova | | 2019-01-24 14:17:15 | View |
Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network
Maud Irène Tenaillon, Khawla Sedikki, Maeva Mollion, Martine Le Guilloux, Elodie Marchadier, Adrienne Ressayre, Christine Dillmann
https://doi.org/10.1101/461947
Early and late flowering gene expression patterns in maize
Recommended by Tanja Pyhäjärvi based on reviews by Laura Shannon and 2 anonymous reviewers
Artificial selection experiments are key experiments in evolutionary biology. The demonstration that application of selective pressure across multiple generations results in heritable phenotypic changes is a tangible and reproducible proof of the evolution by natural selection.
Artificial selection experiments are used to evaluate the joint effects of selection on multiple traits, their genetic covariances and differences in responses in different environments. Most studies on artificial selection experiments report and base their analyses on phenotypic changes [1]. More recently, changes in allele frequency and other patterns of molecular genetic diversity have been used to identify genomic locations where selection has had an effect. However, so far the changes in gene expression have not been in the focus of artificial selection experiment studies (see [2] for an example though).
In plants, one of the most famous artificial selection experiments is the Illinois Corn Experiment where maize (Zea mays) is selected for oil and protein content [3], but in addition, similar experiments have been conducted also for other traits in maize. In Saclay divergent selection experiment [4] two maize inbred lines (F252 and MBS847) have been selected for early and late flowering for 13 generations, resulting in two week difference in flowering time.
In ”Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network ” [5] Maud Tenaillon and her coworkers study the gene expression differences among these two independently selected maize populations. Their experiments cover two years in field conditions and they use samples of shoot apical meristem at three different developmental stages: vegetative, transitioning and reproductive. They use RNA-seq transcriptome level differences and qRT-PCR for gene expression pattern investigation. The work is continuation to earlier genetic and phenotypic studies on the same material [4, 6].
The reviewers and I agree that dataset is unique and its major benefit is that it has been obtained from field conditions similar to those that species may face under natural setting during selection. Their tissue sampling is supported by flowering time phenotypic observations and covers the developmental transition stage, making a good effort to identify key transcriptional and phenotypic changes and their timing affected by selection.
Tenaillon et al. [5] identify more than 2000 genes that are differentially expressed among early and late flowering populations. Expectedly, they are enriched for known flowering time genes. As they point out, differential expression of thousands of genes does not mean that they all were independently affected by selection, but rather that the whole transcriptional network has shifted, possibly due to just few upstream or hub-genes. Also, the year-to-year variation had smaller effect in gene expression compared to developmental stage or genetic background, possibly indicating selection for stability across environmental fluctuation for such an important phenotype as flowering time.
Another noteworthy observation is that they find convergent patterns of transcriptional changes among the two selected lines. 115 genes expression patterns are shifted due to selection in both genetic backgrounds. This convergent pattern can be a result of either selection on standing variation or de novo mutations. The data does not allow testing which process is underlying the observed convergence. However, their results show that this is an interesting future question that can be addressed using genotype and gene expression data from the same ancestral and derived material and possibly their hybrids.
References
[1] Hill, W. G., & Caballero, A. (1992). Artificial selection experiments. Annual Review of Ecology and Systematics, 23(1), 287-310. doi: 10.1146/annurev.es.23.110192.001443
[2] Konczal, M., Babik, W., Radwan, J., Sadowska, E. T., & Koteja, P. (2015). Initial molecular-level response to artificial selection for increased aerobic metabolism occurs primarily through changes in gene expression. Molecular biology and evolution, 32(6), 1461-1473. doi: 10.1093/molbev/msv038
[3] Moose, S. P., Dudley, J. W., & Rocheford, T. R. (2004). Maize selection passes the century mark: a unique resource for 21st century genomics. Trends in plant science, 9(7), 358-364. doi: 10.1016/j.tplants.2004.05.005
[4] Durand, E., Tenaillon, M. I., Ridel, C., Coubriche, D., Jamin, P., Jouanne, S., Ressayre, A., Charcosset, A. and Dillmann, C. (2010). Standing variation and new mutations both contribute to a fast response to selection for flowering time in maize inbreds. BMC evolutionary biology, 10(1), 2. doi: 10.1186/1471-2148-10-2
[5] Tenaillon, M. I., Seddiki, K., Mollion, M., Le Guilloux, M., Marchadier, E., Ressayre, A. and Dillmann C. (2019). Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network. BioRxiv, 461947 ver. 5 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/461947
[6] Durand, E., Tenaillon, M. I., Raffoux, X., Thépot, S., Falque, M., Jamin, P., Bourgais A., Ressayre, A. and Dillmann, C. (2015). Dearth of polymorphism associated with a sustained response to selection for flowering time in maize. BMC evolutionary biology, 15(1), 103. doi: 10.1186/s12862-015-0382-5
| Transcriptomic response to divergent selection for flowering time in maize reveals convergence and key players of the underlying gene regulatory network | Maud Irène Tenaillon, Khawla Sedikki, Maeva Mollion, Martine Le Guilloux, Elodie Marchadier, Adrienne Ressayre, Christine Dillmann | <p>Artificial selection experiments are designed to investigate phenotypic evolution of complex traits and its genetic basis. Here we focused on flowering time, a trait of key importance for plant adaptation and life-cycle shifts. We undertook div... |  | Adaptation, Experimental Evolution, Expression Studies, Quantitative Genetics | Tanja Pyhäjärvi | | 2018-11-23 11:57:35 | View |
When sinks become sources: adaptive colonization in asexuals
Florian Lavigne, Guillaume Martin, Yoann Anciaux, Julien Papaïx, Lionel Roques
https://doi.org/10.1101/433235
Fisher to the rescue
Recommended by François Blanquart and Florence Débarre based on reviews by 3 anonymous reviewers
The ability of a population to adapt to a new niche is an important phenomenon in evolutionary biology. The colonisation of a new volcanic island by plant species; the colonisation of a host treated by antibiotics by a-resistant strain; the Ebola virus transmitting from bats to humans and spreading epidemically in Western Africa, are all examples of a population invading a new niche, adapting and eventually establishing in this new environment.
Adaptation to a new niche can be studied using source-sink models. In the original environment —the “source”—, the population enjoys a positive growth-rate and is self-sustaining, while in the new environment —the “sink”— the population has a negative growth rate and is able to sustain only by the continuous influx of migrants from the source. Understanding the dynamics of adaptation to the sink environment is challenging from a theoretical standpoint, because it requires modelling the demography of the sink as well as the transient dynamics of adaptation. Moreover, local selection in the sink and immigration from the source create distributions of genotypes that complicate the use of many common mathematical approaches.
In their paper, Lavigne et al. [1], develop a new deterministic model of adaptation to a harsh sink environment in an asexual species. The fitness of an individual is maximal when a number of phenotypes are tuned to an optimal value, and declines monotonously as phenotypes are further away from this optimum. This model —called Fisher’s Geometric Model— generates a GxE interaction for fitness because the phenotypic optimum in the sink environment is distinct from that in the source environment [2]. The authors circumvent mathematical difficulties by developing an original approach based on tracking the deterministic dynamics of the cumulant generating function of the fitness distribution in the sink. They derive a number of important results on the dynamics of adaptation to the sink:
From the point where immigration from the source to the sink starts, four phases of adaptation are observed. After a short transient phase (phase 1), a migration-selection balance is reached in the sink (phase 2). After a while, thanks to the immigration of rare adapted migrants and mutation in the sink, a small fraction of the sink population exhibits a close-to-optimal phenotype. This small adapted fraction grows in frequency and mean fitness rapidly increases in the sink (phase 3). Finally, the population settles around the sink optimum (phase 4) and, hurray, the sink is now a source! Interestingly, in this model the evolutionary dynamics do not depend on the immigration rate. In other words, adaptation will proceed at the same rate regardless of how many immigrants invade the sink. This is because the impact of immigration on adaptation depends on the rate of immigration relative to the sink density. This ratio is actually independent of immigration in a model where the sink is initially empty, migration from the sink back to the source is negligible and without density-dependence in the sink. In this model, mutation is a double-edged sword. Adapted phenotypes emerge from new mutations, and under this effect alone a higher mutation rate would translate into a shorter time to establishment in the sink. However, mutations may also have deleterious effects by displacing the phenotype away from the optimum. This mutation load will be greater when individuals need to simultaneously tune a large number of phenotypes. As a consequence of these two effects of mutations, time to establishment is minimal for an intermediate mutation rate. This result emerges from Fisher’s Geometric Model, but may hold more generally for biologically plausible fitness landscapes where mutations generates both beneficial (allowing adaptation to the sink) and deleterious genotypes. Lastly, in Fisher’s Geometric Model, the time to establishment increases superlinearly with harshness of the sink when the sink is too harsh, and establishment may occur only after a very long time. In these harsh sinks, the adapted genotypes are very few and increase very slowly in frequency, making the second phase of adaptation much longer. Thus, and as a direct consequence of Fisher’s Geometric Model, adding a “stepping stone” intermediate environment would allow faster adaptation to the extreme environment.
In conclusion, this theoretical work presents a method based on Fisher’s Geometric Model and the use of cumulant generating functions to resolve some aspects of adaptation to a sink environment. It generates a number of theoretical predictions for the adaptive colonisation of a sink by an asexual species with some standing genetic variation. It will be a fascinating task to examine whether these predictions hold in experimental evolution systems: will we observe the four phases of the dynamics of mean fitness in the sink environment? Will the rate of adaptation indeed be independent of the immigration rate? Is there an optimal rate of mutation for adaptation to the sink? Such critical tests of the theory will greatly improve our understanding of adaptation to novel environments.
References
[1] Lavigne, F., Martin, G., Anciaux, Y., Papaïx, J., and Roques, L. (2019). When sinks become sources: adaptive colonization in asexuals. bioRxiv, 433235, ver. 5 peer-reviewed and recommended by PCI Evolutionary Biology. doi: 10.1101/433235
[2] Martin, G., and Lenormand, T. (2006). A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species. Evolution, 60, 893-907. doi: 10.1111/j.0014-3820.2006.tb01169.x
| When sinks become sources: adaptive colonization in asexuals | Florian Lavigne, Guillaume Martin, Yoann Anciaux, Julien Papaïx, Lionel Roques | <p>The successful establishment of a population into a new empty habitat outside of its initial niche is a phenomenon akin to evolutionary rescue in the presence of immigration. It underlies a wide range of processes, such as biological invasions ... |  | Adaptation, Evolutionary Applications, Evolutionary Dynamics, Evolutionary Ecology | François Blanquart | | 2018-10-03 20:59:16 | View |
Strong habitat and weak genetic effects shape the lifetime reproductive success in a wild clownfish population
Océane C. Salles, Glenn R. Almany, Michael L. Berumen, Geoffrey P. Jones, Pablo Saenz-Agudelo, Maya Srinivasan, Simon Thorrold, Benoit Pujol, Serge Planes
https://doi.org/10.5281/zenodo.3476529
Habitat variation of wild clownfish population shapes selfrecruitment more than genetic effects
Recommended by Philip Munday based on reviews by Juan Diego Gaitan-Espitia and Loeske Kruuk
Estimating the genetic and environmental components of variation in reproductive success is crucial to understanding the adaptive potential of populations to environmental change. To date, the heritability of lifetime reproductive success (fitness) has been estimated in a handful of wild animal population, mostly in mammals and birds, but has never been estimated for a marine species. The primary reason that such estimates are lacking in marine species is that most marine organisms have a dispersive larval phase, making it extraordinarily difficult to track the fate of offspring from one generation to the next.
In this study, Salles et al. [1] use an unprecedented 10 year data set for a wild population of orange clownfish (Amphiprion percula) to estimate the environmental, maternal and additive genetic components of life time reproductive success for the self-recruiting portion of the local population. Previous studies show that over 50% of juvenile clownfish recruiting to the population of clownfish at Kimbe Island (Kimbe Bay, PNG) are natal to the population. In other words, >50% of the juveniles recruiting to the population at Kimbe Island are offspring of parents from Kimbe Island. The identity and location of every adult clownfish in the Kimbe Island population was tracked over 10 years. At the same time newly recruiting juveniles were collected at regular intervals (biennially) and their parentage assigned with high confidence by 22 polymorphic microsatellite loci. Salles et al. then used a pedigree comprising 1735 individuals from up to 5 generations of clownfish at Kimbe Island to assess the contribution of every breeding pair of clownfish to self-recruitment within the local population. Because clownfish are site attached and live in close association with a host sea anemone, it was also possible to examine the contribution of reef location and host anemones species (either Heteractis magnifica or Stichodactyla gigantea) to reproductive success within the local population.
The study found that breeders from the eastern side of Kimbe Island, and mostly inhabiting S. gigantea sea anemones, produced more juveniles that recruited to the local population than breeders from other location around the island, or inhabiting H. magnifica. In fact, host anemone species and geographic location explained about 97% of the variance in reproductive success within the local population (i.e. excluding successful recruitment to other populations). By contrast, maternal and additive genetic effects explained only 1.9% and 1.3% of the variance, respectively. In other words, reef location and the species of host anemone inhabited had an overwhelming influence on the long-term contribution of breeding pairs of clownfish to replenishment of the local population. This overwhelming effect of the local habitat on reproductive success means that the population is potentially susceptible to rapid environmental changes - for example if S. giganta sea anemones are disproportionately susceptible to global warming, or reef habitats on the eastern side of the island are more susceptible to disturbance. By contrast, the small component of additive genetic variance in local reproductive success translated into low heritability and evolvability of lifetime reproductive success within the local population, as predicted by theory [2] and observed in some terrestrial species. Consequently, fitness would evolve slowly to environmental change.
Establishing the components of variation in fitness in a wild population of marine fishes is an astonishing achievement, made possible by the unprecedented long-term individual-level monitoring of the entire population of clownfish at Kimbe Island. A next step in this research would be to include other clownfish populations that are demographically and genetically connected to the Kimbe Island population through larval dispersal. It would be intriguing to establish the environmental, maternal and additive genetic components of reproductive success in the dispersing part of the Kimbe Island population, to see if this potentially differs among breeders who contribute more or less to replenishment within the local population.
References
[1] Salles, O. C., Almany, G. R., Berumen, M.L., Jones, G. P., Saenz-Agudelo, P., Srinivasan, M., Thorrold, S. R., Pujol, B., Planes, S. (2019). Strong habitat and weak genetic effects shape the lifetime reproductive success in a wild clownfish population. Zenodo, 3476529, ver. 3 peer-reviewed and recommended by Peer Community In Evolutionary Biology. doi: 10.5281/zenodo.3476529
[2] Fisher, R.A. (1930). The genetical theory of natural selection. Clarendon Press, Oxford, U.K.
| Strong habitat and weak genetic effects shape the lifetime reproductive success in a wild clownfish population | Océane C. Salles, Glenn R. Almany, Michael L. Berumen, Geoffrey P. Jones, Pablo Saenz-Agudelo, Maya Srinivasan, Simon Thorrold, Benoit Pujol, Serge Planes | <p>Lifetime reproductive success (LRS), the number of offspring an individual contributes to the next generation, is of fundamental importance in ecology and evolutionary biology. LRS may be influenced by environmental, maternal and additive genet... |  | Adaptation, Evolutionary Ecology, Life History, Quantitative Genetics | Philip Munday | | 2018-10-01 09:00:53 | View |
Evolution of selfing & lifespan 2.0
Recommended by Thomas Bataillon based on reviews by 2 anonymous reviewers
Flowering plants display a staggering diversity of both mating systems and life histories, ranging from almost exclusively selfers to obligate outcrossers, very short-lived annual herbs to super long lived trees. One pervasive pattern that has attracted considerable attention is the tight correlation that is found between mating systems and lifespan [1]. Until recently, theoretical explanations for these patterns have relied on static models exploring the consequences of several non-mutually exclusive important process: levels of inbreeding depression and ability to successfully were center stage. This make sense: successful colonization after long‐distance dispersal is far more likely to happen for self‐compatible than for self‐incompatible individuals in a sexually reproducing species. Furthermore, inbreeding depression (essentially a genetically driven phenomenon) and reproductive insurance are expected to shape the evolution of both mating system and lifespan.
But modelling jointly several processes and how their interplay to shape the evolution of a trait is challenging enough so models for the evolution of mating system tend invariably – for mathematical convenience and tractability – to fix lifespan [2].
However, comparative analysis of between species variations that map traits transitions among sister species in phylogenetic trees reveals a pervasive pattern: frequent transitions from a state outcrossing perennial to selfing annuals. This beg the question: is one transition triggering the other and if so, what comes first or are these transitions happening together?
In this work, Lesaffre and Billiard use a very sophisticated machinery developed by Kirkpatrick et al. [3] and consider a general class of so-called modifiers models [4]. They study jointly the evolution of life span and mating system. They do so by using models where different life stages are tracked with life stage having some (fixed for once) amount of inbreeding depression. Their paper is technically demanding, mixing analytics and computer simulations, and along the way generates several important findings that are expected to stimulate further empirical and theoretical studies: (1) pure selfing versus pure outcrossing is the expected stable evolutionary outcomes (despite observation that mixed mating systems can be regularly met in nature), (2) increasing life-span drastically reduces the scope for the evolution of selfing, conversely (3) transition to selfing will also select for shorter life span as a way to mitigate the cumulative effects of inbreeding depression on adult life stages.
As usual there is room for future work, in particular the authors’ model assumes fixed inbreeding depression in the different life stages and this highlights the need for models that explore how inbreeding depression, a pivotal quantity in these models, can itself be molded by both mating system and lifespan. A third-generation of models should be “soon” on the way!
References
[1] Grossenbacher D, Briscoe Runquist R, Goldberg EE, and Brandvain Y. (2015) Geographic range size is predicted by plant mating system. Ecology Letters 18, 706–713. doi: 10.1111/ele.12449
[2] Morgan MT, Schoen DJ, and Bataillon T. (1997) The evolution of self-fertilization in perennials. The American Naturalist 150, 618–638. doi: 10.1086/286085
[3] Kirkpatrick M, Johnson T, and Barton N. (2002) General models of multilocus evolution. Genetics 161, 1727–1750.
[4] Lesaffre, T, and Billiard S. (2019) The joint evolution of lifespan and self-fertilisation. bioRxiv, 420877, ver. 3 peer-reviewed and recommended by PCI Evol Biol. doi: 10.1101/420877
| The joint evolution of lifespan and self-fertilisation | Thomas Lesaffre, Sylvain Billiard | <p>In Angiosperms, there exists a strong association between mating system and lifespan. Most self-fertilising species are short-lived and most predominant or obligate outcrossers are long-lived. This association is generally explained by the infl... |  | Evolutionary Theory, Life History, Reproduction and Sex | Thomas Bataillon | | 2018-09-19 10:03:51 | View |







